INTRODUCCIÓN
El síndrome de Kearns Sayre (SKS) es un tipo de enfermedad mitocondrial cuyos criterios diagnósticos incluyen la presencia de oftalmoplejía externa progresiva (OEP), retinitis pigmentaria y comienzo de los síntomas antes de los 20 años1. Además, debe cumplir uno de los siguientes criterios menores: bloqueo de la conducción cardíaca, síndrome cerebeloso o proteinorraquia mayor de 100 mg/dl. Además de estas manifestaciones pueden encontrarse otros síntomas no específicos, como talla baja, sordera neurosensorial, retraso mental y anormalidades endocrinas (diabetes mellitus, hipoparatiroidismo y déficit de hormona de crecimiento). Se consideran SKS incompletos aquellos que presentan OEP pero falta alguno de los criterios clínicos o se completa la sintomatología muchos años después de su inicio y muestran las delecciones típicas de la enfermedad2.
El curso de la enfermedad es progresivo, dependiendo en gran medida la supervivencia de las alteraciones en la conducción cardíaca.
CASO CLINICO
Se trataba de una paciente de 41 años de edad sin alergias conocidas ni hábitos tóxicos. Entre sus antecedentes destacaba una apendicectomía. Su madre y su abuela no padecían enfermedades de interés, y tenía una hermana y dos hijas de corta edad sanas.
A los 17 años comenzó con un cuadro de caída del párpado derecho y más tarde del izquierdo, siendo diagnosticada de oftalmoplejía externa progresiva (OEP). En la biopsia muscular se observaba la presencia de fibras rojo rasgadas o ragged red (FRR). Presentaba un estudio electroneurográfico y electromiográfico normal.
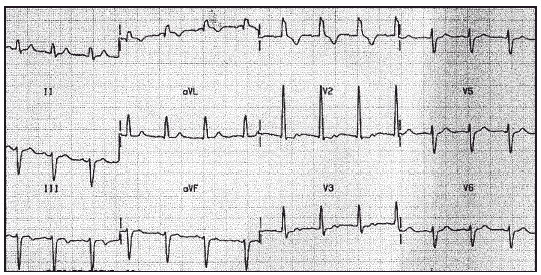
Las determinaciones de creatinfosfocinasa (CPK), aldolasa y anticuerpos anti-RACA eran normales. El fondo de ojo no evidenciaba alteraciones. El ecocardiograma era compatible con la normalidad. En el electrocardiograma (ECG) se observaba un bloqueo bifascicular (fig. 1). Se encontraba en tratamiento con L-carnitina base (Carnicor®) desde 1994.
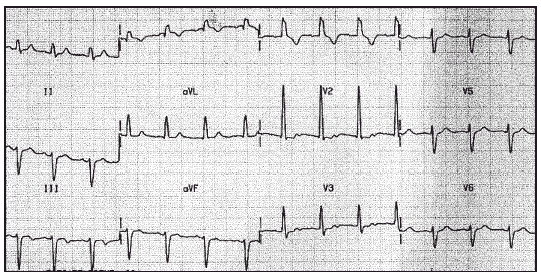
Figura 1. ECG con 12 derivaciones. Ritmo sinusal. Bloqueo bifascicular.
En febrero de 2001 acudió a nuestro hospital aquejando un cuadro clínico de 3-4 días de evolución de mareo y precordalgia inespecífica coincidentes con el ejercicio. No refería sensación disneica ni síncope.
La exploración física mostraba una paciente consciente y orientada, con una puntuación de la escala de Glasgow de 15. Estaba normocoloreada y normohidratada. No se apreciaba ingurgitación yugular. La auscultación cardíaca reflejaba unos tonos bradicárdicos, con un soplo sistólico de regurgitación mitral. La auscultación pulmonar era normal. El abdomen y las extremidades inferiores no presentaban alteraciones de interés. En la exploración neurológica destacaba una ptosis bilateral con limitación de todos los movimientos oculares, sin que se apreciase otra focalidad.
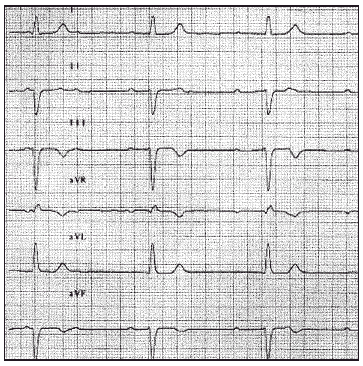
Entre las exploraciones complementarias destacaba el ECG, en el cual se apreciaba un bloqueo auriculoventricular completo con respuesta ventricular a 37 lat/min (fig. 2). El ecocardiograma mostraba una contractilidad global y segmentaria conservada, observando dos jets de regurgitación mitral (II/IV) e insuficiencia tricuspidea (I/IV). En la radiografía de tórax no se apreciaban alteraciones de interés. La analítica revelaba unas enzimas cardíacas normales.
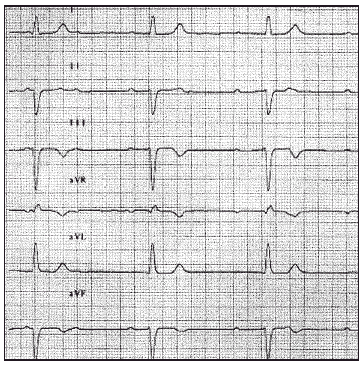
Figura 2. Derivaciones de las extremidades. Bloqueo auriculoventricular completo.
Permaneció inicialmente monitorizada en la unidad de cuidados intensivos sin precisar la colocación de marcapasos provisional urgente al no presentar síntomas o signos de compromiso hemodinámico. Posteriormente, se procedió a la colocación del marcapasos definitivo. La paciente fue dada de alta de la unidad de cuidados intensivos asintomática, y quedó pendiente de un nuevo estudio oftalmológico.
DISCUSIÓN
Las enfermedades mitocondriales constituyen un grupo muy heterogéneo de trastornos en los que existe alguna alteración en la cadena respiratoria mitocondrial. En la biogénesis mitocondrial interviene el ADN nuclear (ADNnucl) y el ADN mitocondrial (ADNmit). Los defectos que se localizan en el ADNnucl se heredan por un patrón mendeliano y los que se localizan a en el ADNmit se heredan siguiendo el patrón de "herencia materna" en la cual todos los descendientes de una mujer afectada presentarán la enfermedad pero solamente las hijas lo transmitirán a su vez a la siguiente generación. Esto se debe a que prácticamente la totalidad del ADNmit lo aporta el óvulo, mientras que el escasísimo número de moléculas del espermatozoide sufriría un proceso de eliminación o bien no tiene ninguna repercusión sobre el fenotipo. La expresión fenotípica de estas mutaciones es variable, debido a que con frecuencia coexisten moléculas de ADNmit normales con otras mutadas (segregación mitótica), y es necesario que se alcance una determinada proporción de genoma mutado no funcional para que la enfermedad se exprese clínicamente (efecto umbral). Esta proporción es distinta para cada tejido e incluso puede ser variable en el tiempo en función de sus necesidades energéticas. Así, órganos como el corazón, el cerebro o el músculo esquelético (en especial la musculatura ocular) son particularmente vulnerables.
Pero también existen una serie de procesos esporádicos, como la oftalmoplejía externa progresiva esporádica con FRR y el síndrome de Kearns Sayre, no sujetos a historia familiar. Este fenómeno podría explicarse por dos mecanismos teóricos: se trata de mutaciones de novo que se producen en el cigoto y que afectan a células somáticas y no a las germinales, o los óvulos con delecciones en el ADNmit pierden su funcionalidad y aunque fueran fertilizados nunca llegarían a completar el desarrollo embrionario3. Creemos que éste sería nuestro caso, dada la ausencia de enfermedad familiar.
En 1958 Thomas P. Kearns y George P. Sayre describieron por primera vez este síndrome, que lleva su nombre. Tradicionalmente la tríada clínica del SKS incluía oftalmoplejía externa progresiva, retinitis pigmentaria y comienzo de los síntomas antes de los 20 años. Además debía presentar alguno de los criterios menores. Sin embargo, la secuencia de las manifestaciones clínicas no es constante y los signos y los síntomas, aunque variables, pueden ayudar a identificar formas incompletas de la enfermedad (se conocen en ocasiones como OEP-plus)3. En esta paciente no se había observado enfermedad retiniana en un examen anterior, y al alta había quedado pendiente de una nueva revisión oftalmológica4.
En la biopsia muscular es típica la presencia de las fibras ragged red que corresponden a fibras musculares con acumulación de mitocondrias anormales que en el microscopio óptico dan ese aspecto característico. Casi todos los pacientes afectados de SKS presentan grandes delecciones únicas en el ADNmit. El curso de esta enfermedad es progresivo y la mortalidad está estrechamente relacionada con las alteraciones en la conducción cardíaca5. Las principales manifestaciones de la afección cardíaca son el síncope en un 45% y la muerte súbita en un 23%, así como el fallo cardíaco. En un 20% de los pacientes hay una lenta y progresiva miocardiopatía dilatada6.
El electrocardiograma puede reflejar distintas alteraciones de la conducción cardíaca pero es bien conocido que suele evolucionar hacia el bloqueo auriculoventricular completo, causante de la muerte en un 20% de los enfermos7. En nuestro caso, la paciente, aunque presentaba un bloqueo bifascicular en 1995, no se le sometió a estudio electrofisiológico. Tampoco disponemos de más registros electrocardiográficos hasta que, 6 años después, acude a nuestro hospital con síntomas de intolerancia al ejercicio y presíncope, apreciándose en el electrocardiograma un bloqueo auriculoventricular completo. En el ecocardiograma se apreciaba una ligera insuficiencia mitral y tricuspídea, cuya presencia también ha sido recogida en otros estudios.
Por todo ello, algunos grupos han propuesto la implantación profiláctica de marcapasos definitivo sin realización previa de estudios electrofisiológicos a todos los pacientes diagnosticados de SKS que presenten bloqueos bifasciculares asintomáticos8.
En la actualidad, el American College of Cardiology y la American Heart Association recomiendan la implantación de marcapasos definitivo en los pacientes que presenten bloqueos bifasciculares asintomáticos cuando el estudio electrofisiológico demuestre un intervalo H-V ≥ 100 ms9. Sin embargo, otros grupos consideran que la implantación de marcapasos definitivo está justificada cuando se asocian las alteraciones de la conducción descritas anteriormente, con un intervalo H-V ≥ 60 ms, dado el alto riesgo que existe en estas situaciones de progresión a bloqueo auriculoventricular completo. Asimismo, se recomienda que aquellos pacientes con alteraciones eléctricas y exploración electrofisiológica normal se sometan a un estudio electrocardiográfico trimestral y electrofisiológico anual10.