

La hiperactividad simpática paroxística es una urgencia neurológica potencialmente letal secundaria a múltiples lesiones cerebrales agudas adquiridas. Se caracteriza por rasgos clínicos de aparición cíclica y simultánea, consecuencia de una descarga simpática exacerbada. El diagnóstico es clínico, requiriendo elevados índices de alerta. Actualmente no existen criterios diagnósticos homogéneos que estén ampliamente difundidos y validados. El consenso reciente intenta arrojar luz sobre este oscuro panorama. Su fisiopatología es compleja y aún no ha sido elucidada con certeza; sin embargo, la teoría basada en el modelo excitación-inhibición es la que mejor explica los distintos aspectos de esta entidad, incluyendo la respuesta a la terapia con los fármacos disponibles. Los pilares terapéuticos se asientan sobre el reconocimiento precoz, evitar insultos secundarios y el desencadenamiento de los paroxismos. De ocurrir crisis simpáticas, es que estas se aborten de forma perentoria y que se prevengan. Cuanto más tarde en reconocerse el síndrome, peores serán los resultados.
Paroxysmal sympathetic hyperactivity (PSH) is a potentially life-threatening neurological emergency secondary to multiple acute acquired brain injuries. It is clinically characterized by the cyclic and simultaneous appearance of signs and symptoms secondary to exacerbated sympathetic discharge. The diagnosis is based on the clinical findings, and high alert rates are required. No widely available and validated homogeneous diagnostic criteria have been established to date. There have been recent consensus attempts to shed light on this obscure phenomenon. Its physiopathology is complex and has not been fully clarified. However, the excitation-inhibition model is the theory that best explains the different aspects of this condition, including the response to treatment with the available drugs. The key therapeutic references are the early recognition of the disorder, avoiding secondary injuries and the triggering of paroxysms. Once sympathetic crises occur, they must peremptorily aborted and prevented. of the later the syndrome is recognized, the poorer the patient outcome.
En 1929 Wilder Penfield describe el caso de una mujer de 41 años de edad que súbita y paroxísticamente presenta hipertensión, taquicardia, taquipnea con cambios ocasionales del patrón ventilatorio normal a ritmo de Cheyne-Stoke1. Durante los paroxismos asciende la temperatura corporal, las pupilas se dilatan y contraen intermitentemente, junto a excesivo lagrimeo1. La paciente padecía un colesteatoma del tercer ventrículo. Penfield denomino a estas crisis episódicas «epilepsia autonómica diencefálica», sugiriendo que las mismas obedecían a sobreactividad del sistema nervioso autónomo, tanto simpático como parasimpático1.
Desde su publicación inicial, cuadros clínicos similares han sido descritos en múltiples tipos de lesiones cerebrales graves, con diferentes denominaciones2–6. En ocasiones predominaba el compromiso simpático, en otras lo hacia el componente parasimpático o ambos, confundiendo el panorama sin permitir un estudio adecuado de esta entidad2,6 (tabla 1).
Diferentes nomenclaturas de la hiperactividad simpática paroxística a lo largo del tiempo
| Convulsiones diencefálicas |
| Ataque del troncoencéfalo |
| Desregulación autonómica central |
| Estado hiperadrenérgico |
| Síndrome del cerebro medio |
| Espasmos tónicos descerebrantes |
| Descargas tónicas cerebelares |
| Respuesta simpático adrenal |
| Síndrome de disfunción autonómica |
| Síndrome de desregulación hipotalámica-cerebro medio |
| Disautonomía |
| Hiperpirexia con contracción muscular sostenida |
| Tormenta autonómica |
| Tormenta simpática |
| Inestabilidad autonómica paroxística con distonía |
| Disregulación autonómica paroxística |
| Hiperactividad simpática paroxística |
Recientemente, ha sido acuñado el término «hiperactividad simpática paroxística» (HSP), que resume las principales características del síndrome, el cual sobreviene como consecuencia de sobreactividad del sistema nervioso simpático de manera exclusiva2–5,7,8.
La HSP es una verdadera urgencia neurológica que puede pasar desapercibida si no se tiene en cuenta2–8. Su diagnóstico, principalmente de exclusión, requiere un alto índice de sospecha, ya que de no detectarse y tratarse de manera adecuada se asocia a elevadas tasas de morbimortalidad2–5.
El objetivo principal del presente artículo es brindar de manera sencilla y práctica respuestas a interrogantes planteados para reconocer y manejar esta entidad de la manera más correcta posible.
Interrogante 1. ¿A qué denominamos hiperactividad simpática paroxística?La HSP es el conjunto de signos y síntomas que denotan descarga simpática exacerbada, entre los que se encuentran: taquicardia, hipertensión arterial, taquipnea, hipertermia, sudoración generalizada, posturas motoras anómalas (distonías, rigidez muscular, extensión), desadaptación del ventilador mecánico2–8. Los rasgos clínicos señalados se presentan de manera simultánea2–8. El número de signos o síntomas presentes tiene estrecha correlación con el resultado final4. A mayor número, mayor probabilidad de malos resultados4.
La HSP no es una entidad primaria, se desarrolla siempre como consecuencia de lesiones cerebrales de distinta índole y gravedad2–8.
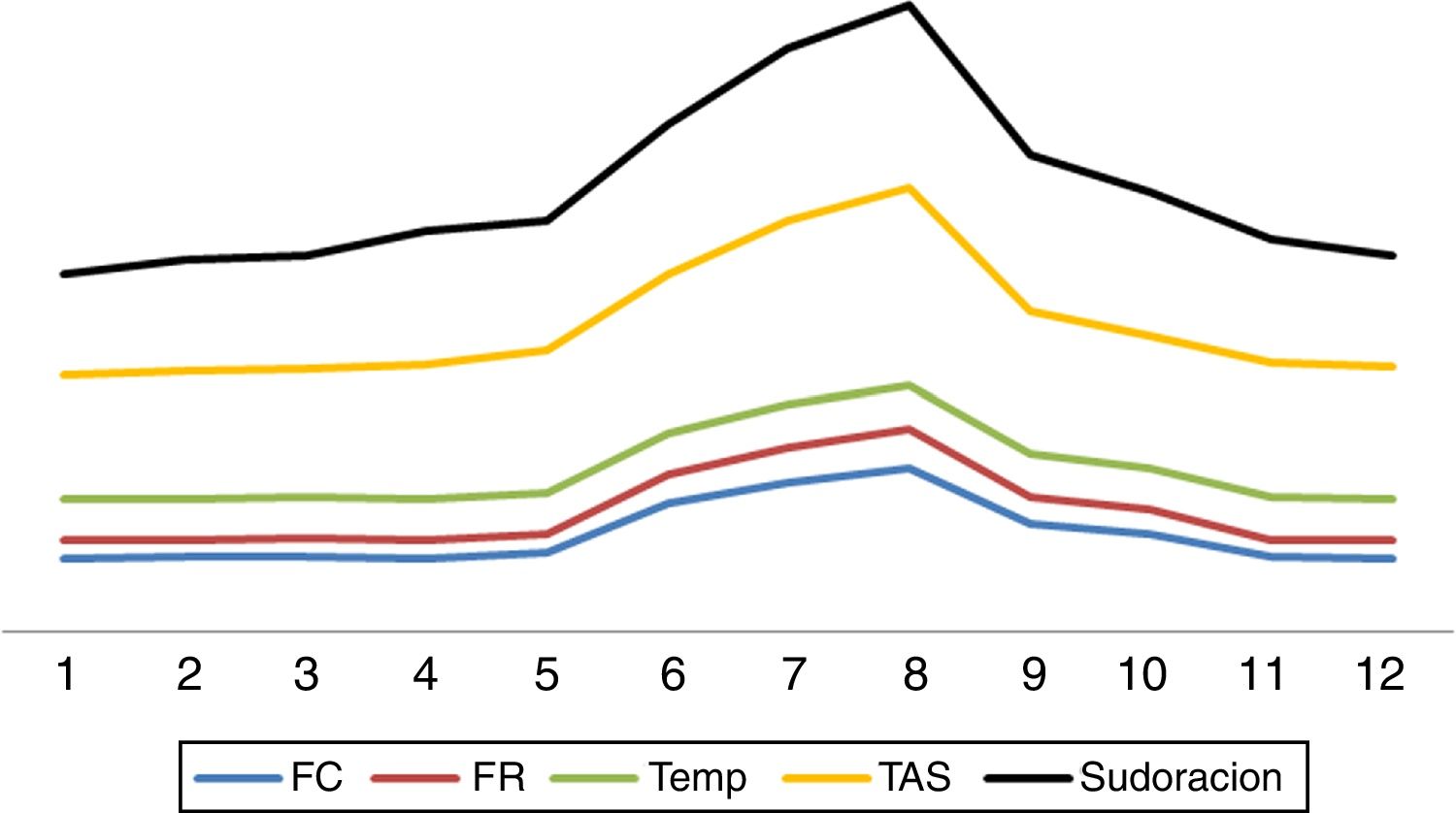
Interrogante 2. ¿Cuáles son las características principales de hiperactividad simpática paroxística?La HSP se caracteriza por presentación abrupta, en crisis cíclicas, ya sea de manera espontánea o ante estímulos como el dolor, baño, aspiración de secreciones, luz, tacto o fisioterapia2–8. Los episodios de hiperactividad simpática pueden aparecer en cualquier momento de la evolución de la entidad que lo motivó, siendo usualmente detectado después de la primera semana, coincidiendo con la disminución o cese de sedoanalgesia profunda2–8 (fig. 1). Los estudios señalan que la mayoría de las veces el diagnóstico se efectúa con un retraso de una semana en relación con la admisión8. Los paroxismos se presentan entre 3 y 5 veces por día, con una duración media de 30min2–8.

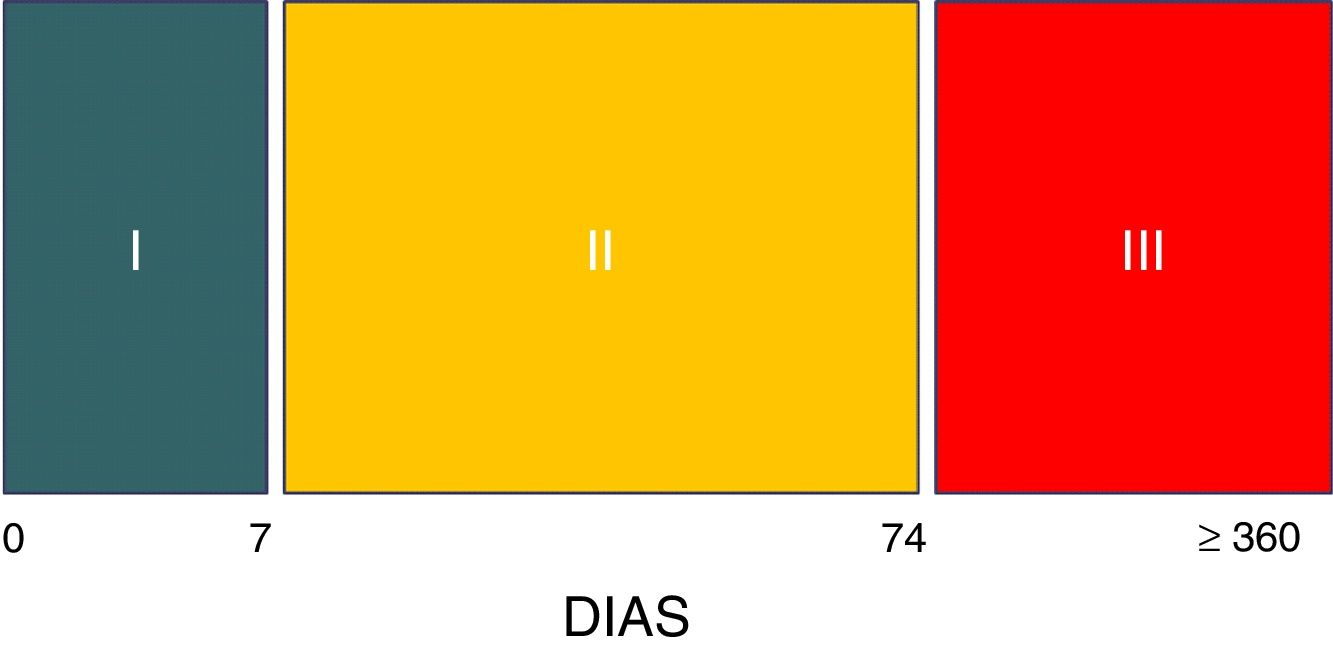
Tres son las etapas evolutivas del síndrome8. La fase i hiperaguda comprende la primera semana durante la cual la lesión cerebral se encuentra en su máxima expresión, generalmente inestable, con altos niveles de intensidad terapéutica, entre los que se encuentran sedación y analgesia generalmente profunda, con lo cual el diagnóstico es imposible de efectuar, salvo que por alguna razón se realice un test del despertar o el individuo accidentalmente salga del plano anestésico8. En la fase ii el síndrome, con las características señaladas, se expresa en su totalidad. Esta etapa se extiende por término medio hasta los 2 meses y medio poslesión (día 74).
Marca el fin de esta fase el cese de los episodios de sudoración8. La fase iii transcurre durante el periodo de rehabilitación y puede extenderse durante años, aunque las crisis habitualmente son de menor frecuencia, intensidad y duración8 (fig. 2).

Fases evolutivas de la hiperactividad simpática paroxística
Fuente: Hughes y Rabinstein8.
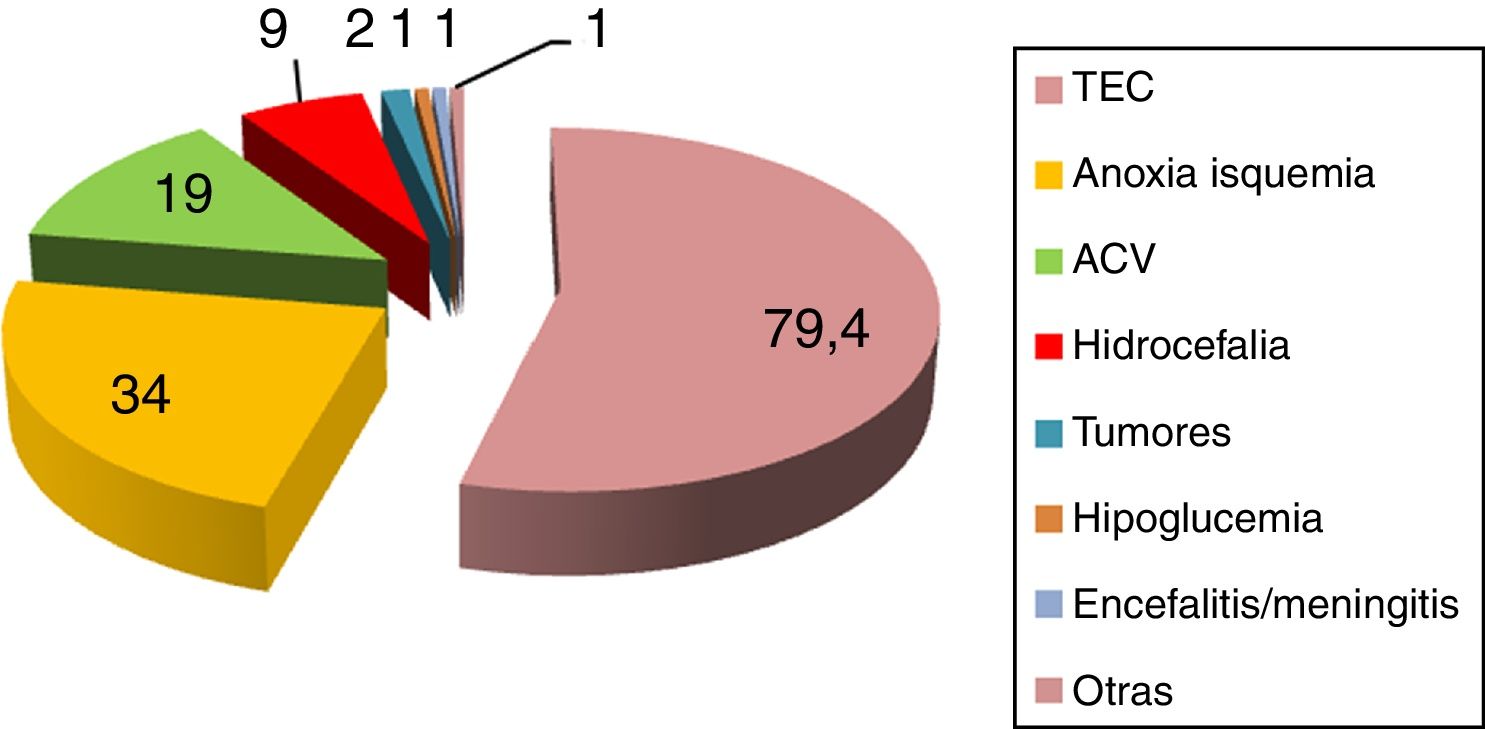
La incidencia publicada de HSP oscila entre el 8% y el 33%2–8, dependiendo de la serie y etiología subyacente. Se desarrolla tanto en adultos como en niños2–8, sin respetar edad ni sexo. Predisponen al desarrollo de HSP diversas etiologías, siendo la más frecuente el traumatismo craneoencefálico grave (80%) en su forma clínica de lesión axonal difusa9–14, seguido por encefalopatía anóxica-isquémica posparada cardiaca (10%) y ataque cerebrovascular (ACV) (5,5%)2–8. En referencia al ACV predomina la hemorragia intraparenquimatosa espontánea en núcleos basales, talámo y vermis cerebeloso, con o sin volcado ventricular15,16. Se han descrito, asimismo, cuadros de HSP en pacientes con hemorragia subaracnoidea de mal grado17,18, ACV isquémico, trombosis venosa cerebral, encefalitis y embolia grasa cerebral2–5,19,20 (fig. 3).

Entidades que predisponen al desarrollo de hiperactividad simpática paroxística.
ACV: accidente cerebrovascular; TCE: traumatismo craneoencefálico.
Fuente: Perkes et al.2.
El actor principal es el sistema nervioso autónomo (SNA) o neurovegetativo, encargado de regular acciones involuntarias, a través del funcionamiento de sus 3 componentes principales: simpático, parasimpático y entérico21. El SNA es esencialmente eferente. Recibe información del medio interno, glándulas y vísceras mediante una compleja red situada en la médula espinal, troncoencéfalo, estructuras diencefálicas, hipotálamo, sistema límbico y ciertas zonas de la corteza cerebral, para luego transmitir el impulso hacia la periferia21. De esa manera regula la frecuencia cardiaca, respiratoria, el diámetro pupilar y de los vasos sanguíneos, la contracción del músculo liso, la salivación, la sudoración, la micción, la función sexual, la secreción de glándulas endocrinas y exocrinas y la digestión21.
El sistema nervioso simpático está conformado por redes ganglionares preaórticas, pre y paravertebrales y su neurotransmisor principal son las catecolaminas, cuya secreción se halla finamente regulada y balanceada por impulsos excitatorios e inhibitorios21. Cuando este equilibrio se altera como consecuencia de lesión cerebral las catecolaminas se liberan excesivamente provocando los rasgos clínicos de la HSP22,23.
Ahora bien, ¿por qué se produce el desequilibrio que ocasiona incremento del nivel de catecolaminas durante los paroxismos simpáticos? La fisiopatología exacta hasta hoy permanece en el terreno de las hipótesis. Inicialmente el síndrome era atribuido a convulsiones o hipertensión endocraneal. Ambas situaciones fueron descartadas, ya que no existe una correlación estrecha entre ambas variables y la presencia del síndrome2–5,24,25. La ausencia de descargas convulsivas en el EEG, durante los paroxismos simpáticos, ha sido claramente objetivada2,24,25. Por otra parte, la elevación de la PIC es más una consecuencia que la causa del síndrome2–5.
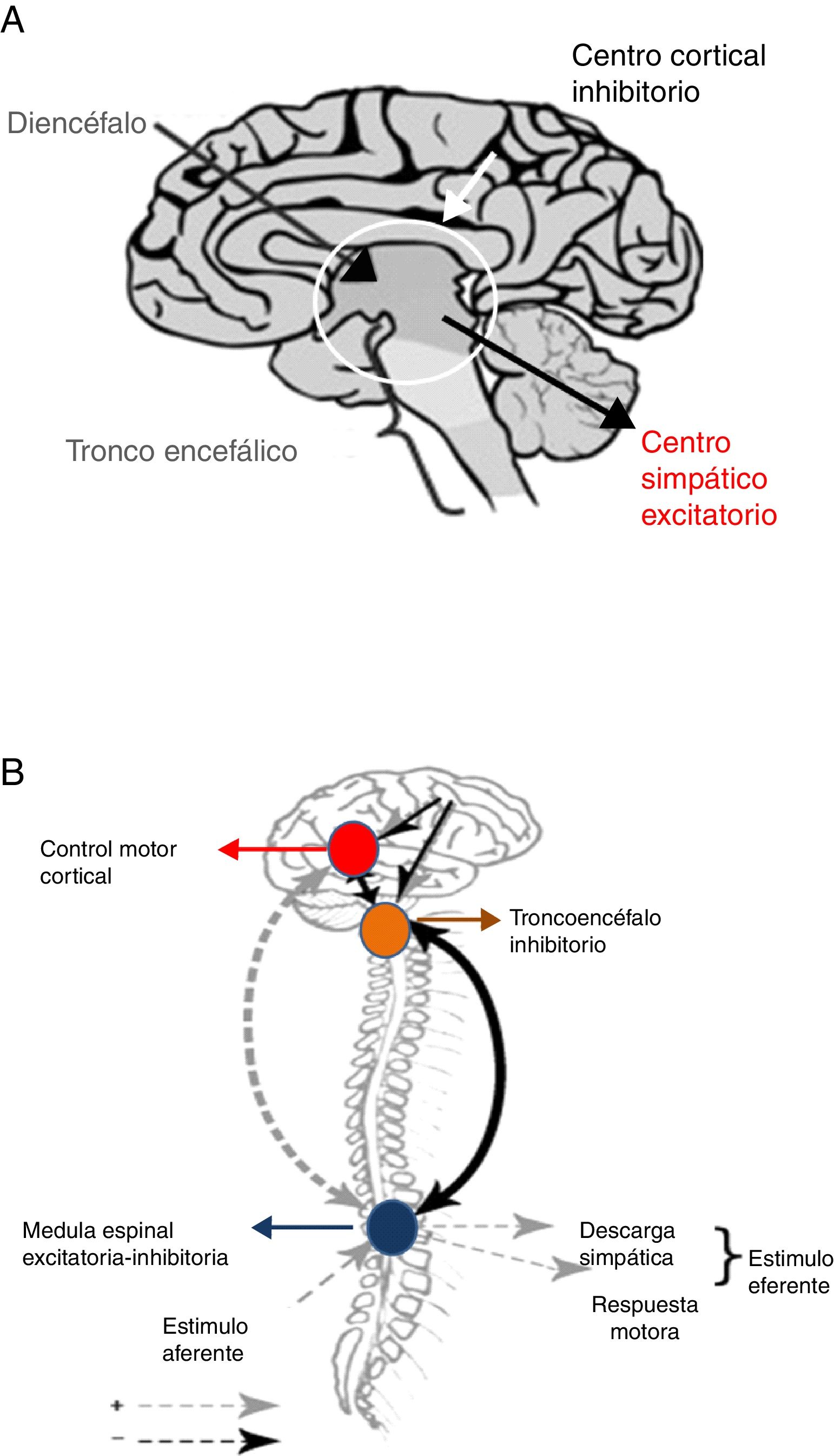
Tras la lesión cerebral se produce, inmediatamente, una respuesta metabólica e inflamatoria con la activación del SNA26, que como consecuencia provoca taquicardia, hipertensión arterial y redistribución del flujo de sangre hacia el cerebro, corazón y glándulas suprarrenales, a fin de asegurar la disponibilidad de oxígeno y preservar procesos fisiológicos en los órganos vitales. El sistema parasimpático intenta restaurar la homeostasis mediante la reducción de los efectos de la hiperactividad simpática. Sin embargo, cuando esta retroalimentación parasimpática falla, el flujo simpático es desinhibido, lo que desencadena hiperactividad y en última instancia HSP. Dos hipótesis intentan explicar este fenómeno24,25 (fig. 4).

Teorías de desconexión: A. Convencional. B. Modelo excitación-inhibición.
Fuente: Baguley25.
La teoría de la desconexión convencional, ya sea estructural (lesión anatómica) o funcional (disbalance en la liberación de neurotransmisores) señala que centros excitatorios simpáticos localizados en el diencéfalo y la parte superior del tronco son liberados del control superior córtico-subcortical24.
El modelo de relación excitación-inhibición propone que los centros localizados en el tronco cerebral y diencéfalo son inhibitorios por naturaleza. De esa forma, limitan la amplificación y sensibilización de la información sensorial aferente proveniente de la médula espinal24,25. En este modelo la HSP es denominada alodinia, término que designa un proceso de sensibilización que ocurre en el cuerno dorsal de la médula espinal, mediante el cual un estímulo no doloroso es percibido como doloroso, entre los que se incluyen: aspiración de secreciones, rotaciones corporales, baño, estreñimiento y retención urinaria24,25. Al mismo tiempo, los estímulos dolorosos se magnifican en intensidad. Adicionalmente, el modelo de relación excitación-inhibición contribuye a explicar el desencadenamiento de HSP ante estímulos ambientales, así como la respuesta a los fármacos utilizados para controlar esta entidad24,25.
Interrogante 5. ¿Cómo efectuar un diagnóstico correcto?Es necesario un alto índice de sospecha, sobre todo cuando en el contexto de una lesión cerebral adquirida, en el período de desescalada terapéutica, «despertar del coma», o en la fase de rehabilitación los individuos presentan episodios paroxísticos, simultáneos y transitorios de hiperactividad simpática2,3,6–8,27.
Los signos y síntomas predominantes son: taquicardia (98%), hipertensión (72%), diaforesis excesiva (79%), fiebre sin foco (79%), taquipnea (85%), postura extensora (38%), distonías (38%), rigidez o espasticidad (44%), mientras que se presentan menos frecuentemente: dilatación pupilar intermitente, disminución del nivel de conciencia, piloroerección, excitación y desadaptación a la ventilación mecánica2,8.
No existe estudio complementario alguno que ayude a establecer el diagnóstico de certeza de HSP, el cual es eminentemente clínico, siendo por ello necesario excluir otras situaciones con rasgos clínicos similares2,3,6–8 (tabla 2).
Entidades que comparten signos o síntomas con hiperactividad simpática paroxística
| Bacteriemia, sepsis |
| Obstrucción de las vías aéreas |
| Hipoxemia |
| Hipercapnia severa |
| Hipoglucemia |
| Convulsiones |
| Deterioro del estado neurológico (hipertensión endocraneal, sangrado, edema, hidrocefalia) |
| Tromboembolismo de pulmón |
| Tormenta tiroidea |
| Infarto agudo de miocardio |
| Abstinencia de alcohol o drogas |
| Síndrome retirada sedación, opioides |
| Síndrome neuroléptico maligno |
| Síndrome serotoninérgico |
| Hipertermia de origen central |
| Hipertermia maligna |
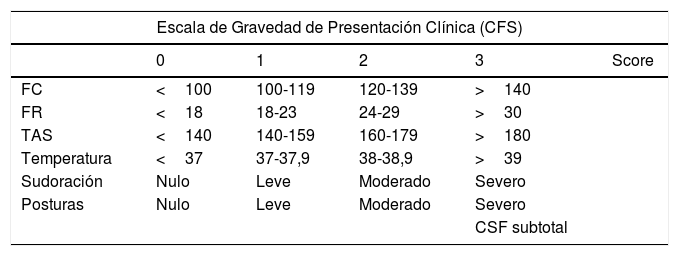
A pesar de ser una entidad conocida desde hace más de 60 años, los criterios necesarios para asegurar el diagnóstico de esta afección no han sido homogéneos ni validados2–8. Diferentes combinaciones y número de signos se han empleado para certificar la existencia de HSP2,28, hasta que, recientemente, un consenso de expertos ha establecido su definición y criterios a emplear29. Tras una revisión sistemática de la literatura se desarrolló una escala basada en la combinación de un score que evalúa la presencia y gravedad de los parámetros clínicos (CSF), cuya puntuación se suma con otra que valora las características de los episodios (frecuencia, duración, mantenimiento en el tiempo, simultaneidad, etc.), denominada herramienta de probabilidad diagnóstica (DLT según sus siglas en inglés)29,30 (tabla 3). La puntuación final obtenida con la suma de estas 2 escalas permite calcular la probabilidad de padecer HSP con mayor precisión, sin embargo su validación se encuentra pendiente.
Escala diagnóstica sugerida por el consenso de expertos
| Escala de Gravedad de Presentación Clínica (CFS) | |||||
|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | Score | |
| FC | <100 | 100-119 | 120-139 | >140 | |
| FR | <18 | 18-23 | 24-29 | >30 | |
| TAS | <140 | 140-159 | 160-179 | >180 | |
| Temperatura | <37 | 37-37,9 | 38-38,9 | >39 | |
| Sudoración | Nulo | Leve | Moderado | Severo | |
| Posturas | Nulo | Leve | Moderado | Severo | |
| CSF subtotal | |||||
| Tipo e intensidad de hipertonicidad durante el episodio | ||
|---|---|---|
| Gravedad de la presentación clínica | Nulo | 0 |
| Leve | 1-6 | |
| Moderado | 7-12 | |
| severo | >13 |
| Herramienta de probabilidad diagnostica (DLT) |
| Simultaneidad del cuadro clínico |
| Presentación paroxística |
| Hiperactividad simpática ante estímulos no dolorosos |
| Persistencia del cuadro clínico >3 días consecutivos |
| Persistencia del cuadro clínico >2 semanas postinjuria |
| Persistencia del cuadro clínico a pesar del tratamiento de diagnósticos alternativos |
| Tratamiento para disminuir rasgos de hiperactividad simpática |
| >2 episodios diarios |
| Ausencia de cuadro clínico parasimpático durante el episodio |
| Ausencia de otras causas responsables del cuadro clínico |
| Antecedentes de lesión cerebral adquirida |
| (1 punto por cada presentación clínica)DLT subtotal |
| Combinación total (CFS+DLT) | ||
|---|---|---|
| Probabilidad de diagnóstico de HSP | Improbable | <8 |
| Posible | 8- 16 | |
| Probable | >17 |
CFS: Clinical Feature Scale. Escala de gravedad de presentación clínica; DLT: Diagnosis likelihood tool. Herramienta de probabilidad diagnóstica; HSP: hiperactividad simpática paroxística.
Fuente: Baguley et al.29.
Si bien no están dilucidados los valores de catecolaminas o de otros ejes hormonales para el diagnóstico de esta condición, un estudio reciente proporciona la primera prueba del incremento de catecolaminas, y en menor medida, de la respuesta adreno-cortical en sujetos con HSP en comparación con un grupo control sin HSP23. Estos resultados proporcionan fundamentos para avalar la actual nomenclatura de este proceso29.
Interrogante 6. ¿Es necesaria la realización de neuroimagen?No es imprescindible para diagnosticar HSP. No obstante, ayuda a mantener un alto índice de sospecha al mostrar en la imagen inicial el tipo de lesión cerebral, focal o difusa, la morfología de la lesión (hemorragia intracerebral, isquemia, contusiones, hematomas extraaxiales, etc.), la localización anatómica y la extensión del daño primario31–33.
Sin duda, en la fase aguda la tomografía computarizada (TC) es de elección. En pacientes más estables, sin necesidad de multimonitorización, puede efectuarse resonancia magnética con o sin tractografía para definir con mayor exactitud el grado de compromiso anatómico, sobre todo de las estructuras anatómicas más vinculadas al desarrollo de HSP34.
Interrogante 7. ¿Cómo abordar el tratamiento de este síndrome?Prevenir la aparición de HSP sería el abordaje ideal; sin embargo, hasta ahora, ninguna medida orientada a este fin se había demostrado exitosa. Recientemente, un estudio retrospectivo, con marcadas limitaciones metodológicas, indica que el empleo de dexmedetomidina, frente a los sedantes tradicionalmente usados (midazolam o propofol), en pacientes con traumatismo craneoencefálico intervenidos quirúrgicamente, podría disminuir la incidencia de HSP35.
Como en cualquier urgencia neurológica, la terapéutica, en primera instancia, debe asegurar la estabilidad cardiorrespiratoria mediante el ABC de la resucitación, conjuntamente con las medidas necesarias para detectar y corregir lesiones secundarias2–5,36–39. Una adecuada nutrición y un balance hidroelectrolítico correcto son medidas auxiliares de suma importancia en los pacientes que padecen HSP39.
Las estrategias farmacológicas vigentes para el manejo específico del HSP se basan en series de casos, la mayoría retrospectivos y no aleatorizados, por lo que la calidad de la evidencia es pobre para evaluar la eficacia de las medidas propuestas. Asimismo, es importante destacar la ausencia de estudios que muestren preferencia de un fármaco sobre otro. No obstante, la experiencia y literatura señalan que generalmente son necesarias «combinaciones de fármacos», así como la necesidad de ir evaluándolos a través del denominado «ensayo-error»3,5,20,36,38.
Para controlar la descarga simpática excesiva o sus consecuencias es importante orientar el tratamiento según la fisiopatología actualmente aceptada de la HSP. De este modo se pueden mitigar los paroxismos mediante 3 vías principales: a) inhibición del flujo simpático central; b) inhibición de procesos sensitivos aferentes (impidiendo el desarrollo de alodinia); y c) bloqueo de la respuesta final del órgano efector3,5,20,36–38.
Actualmente, el tratamiento optimizado (efectividad-reacción adversa-interacciones) (tabla 4) está dirigido a:
- 1)
Abortar crisis o suspensión de los síntomas: conducente al control inmediato del episodio, para evitar que los eventos adversos, tales como sobrecarga cardiaca, arritmias, deshidratación, pérdida muscular, contracciones y recuperación retardada no contribuyan al aumento de la morbilidad. Los fármacos a utilizar se caracterizan por comienzo de acción rápido y vida media corta, y su elección depende del síntoma dominante. Se incluyen aquí la morfina, el propranolol y las benzodiacepinas de acción corta3,5,20,36–38.
- 2)
Prevención de los paroxismos: la terapia se destina a disminuir la frecuencia, duración e intensidad de los síntomas, y se indican en combinación con el grupo anterior. Aquí se incluyen bloqueadores beta no selectivos (propranolol), α2-agonistas (clonidina y en algún grupo de pacientes dexmedetomidina35), bromocriptina, baclofeno, gabapentina y benzodiacepinas de acción prolongada36,37.
- 3)
HSP refractaria: cuando a pesar de las terapias antedichas persisten las descargas simpáticas, con riesgo de desencadenar lesiones cerebrales secundarias, edema cerebral, edema de pulmón, infarto de miocardio, miocarditis catecolaminérgica, inclusive muerte súbita, se recurre a fármacos endovenosos en infusión continua, como benzodiazepinas, propofol, opioides o dexmedetomidina38.
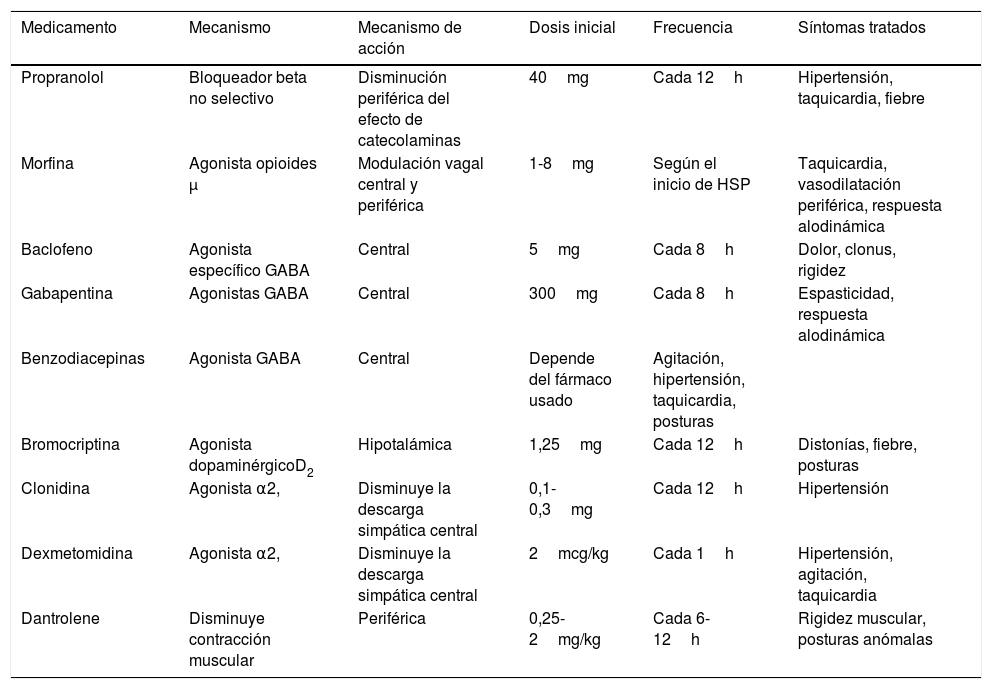
Medicamentos utilizados para el tratamiento de hiperactividad simpática paroxística
| Medicamento | Mecanismo | Mecanismo de acción | Dosis inicial | Frecuencia | Síntomas tratados |
|---|---|---|---|---|---|
| Propranolol | Bloqueador beta no selectivo | Disminución periférica del efecto de catecolaminas | 40mg | Cada 12h | Hipertensión, taquicardia, fiebre |
| Morfina | Agonista opioides μ | Modulación vagal central y periférica | 1-8mg | Según el inicio de HSP | Taquicardia, vasodilatación periférica, respuesta alodinámica |
| Baclofeno | Agonista específico GABA | Central | 5mg | Cada 8h | Dolor, clonus, rigidez |
| Gabapentina | Agonistas GABA | Central | 300mg | Cada 8h | Espasticidad, respuesta alodinámica |
| Benzodiacepinas | Agonista GABA | Central | Depende del fármaco usado | Agitación, hipertensión, taquicardia, posturas | |
| Bromocriptina | Agonista dopaminérgicoD2 | Hipotalámica | 1,25mg | Cada 12h | Distonías, fiebre, posturas |
| Clonidina | Agonista α2, | Disminuye la descarga simpática central | 0,1-0,3mg | Cada 12h | Hipertensión |
| Dexmetomidina | Agonista α2, | Disminuye la descarga simpática central | 2mcg/kg | Cada 1h | Hipertensión, agitación, taquicardia |
| Dantrolene | Disminuye contracción muscular | Periférica | 0,25- 2mg/kg | Cada 6-12h | Rigidez muscular, posturas anómalas |
El manejo clínico eficaz de pacientes con HSP requiere una clara comprensión de las opciones terapéuticas disponibles, su eficacia, dosificación, vida media, vía de administración, interacciones y efectos adversos, por lo que resulta esencial protocolizar el tratamiento. Diferentes algoritmos han sido publicados3,40.
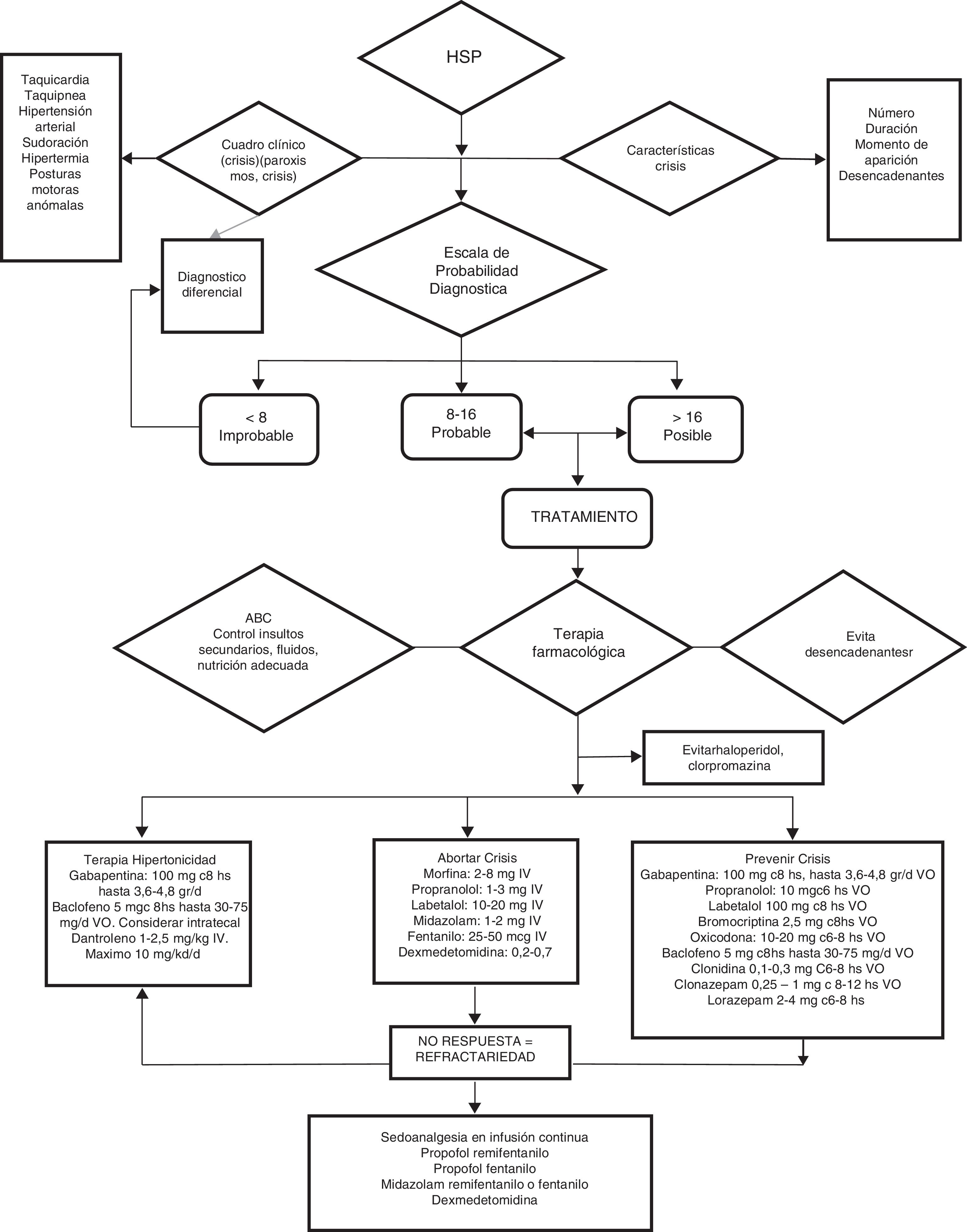
Debido a que, por un lado, no todos los paroxismos son iguales en términos de gravedad, frecuencia y duración, y por otro que los fármacos disponibles no están exentos de toxicidad potencial, creemos importante categorizar los episodios, para emplear la combinación terapéutica con mejor relación riesgo-beneficio. Para ello, recomendamos emplear la puntuación de la escala diagnóstico CFS+DLT y clasificar los pacientes en 4 posibles grupos (fig. 5):

Algoritmo de manejo de la hiperactividad simpática paroxística.
ABC de resucitación: vía aérea permeable, oxigenación y ventilación adecuada, hemodinamia estable.
Observación: haloperidol y clorpromazina deben evitarse por sus efectos antidopaminérgicos que exacerban o agravan el HSP.
d: día; mg: miligramos; mcg: microgramos; g: gramos; IV: intravenoso; VO: vía oral.
Grupo A<8 puntos. Tratamiento dirigido al síntoma dominante.
Grupo B8-16puntos. Terapia sintomática+preventiva.
GrupoC>17puntos. Tratamiento sintomático+preventivo+gabapentina o dantroleno o baclofeno.
Grupo refractario: infusión iv continua de propofol, fentanilo, midazolam o dexmedetomidina.
Interrogante 8. ¿Cuáles son sus consecuencias sin diagnóstico o manejo adecuado?Los episodios de HSP pueden ser intensos, prolongados y pueden repetirse varias veces al día2–6,41. El número de síntomas más que la duración de la HSP es el indicador más importante de gravedad4. La hipertensión arterial, fiebre, hipoxemia, hipercapnia e hiperglucemia pueden generar lesiones cerebrales secundarias, causas principales del pronóstico desfavorable2–6. A su vez la HSP ocasiona un estado hipermetabólico, hipercatabólico e inflamatorio con mayor susceptibilidad a infecciones, sepsis y pérdida de peso asociados a mayor morbilidad, estancia hospitalaria y retraso en la recuperación2–6,20. El incremento marcado y sostenido de catecolaminas predispone al desarrollo de miocardiopatías, edema de pulmón, arritmias, disfunción cardiaca y multisistémica2–8,20. El diagnóstico precoz y el tratamiento optimizado de la HSP son fundamentales para facilitar la recuperación y discapacidad permanente como consecuencia de osificaciones heterotópicas, rigidez espástica, posiciones viciosas y alteraciones profundas de la esfera neurocognitiva2–8,20. El inicio temprano de la terapia específica de los síntomas se piensa que reduce las tasas de complicaciones, estancia en la UCI y facilita la recuperación18.
El retraso diagnóstico-terapéutico de la HSP puede tener consecuencias devastadoras para los programas de recuperación de estos pacientes, sin embargo, si se compara la evolución posrehabilitación de pacientes con HSP y sin HSP, la situación funcional al alta no muestra diferencias estadísticamente significativas medidos por la Functional Independence Measure, Disability Rating Scale y Glasgow Outcome Scale, aunque la probabilidad y el nivel de mejora es superior en aquellos sin HSP42,43.
El curso de recuperación esperado de la lesión cerebral traumática sigue el camino que va desde la atención aguda a la rehabilitación y reinserción a la comunidad. Los efectos de la HSP pueden alterar la trayectoria rehabilitadora y retrasar o prohibir la recuperación máxima43.
Conclusiones y retos futurosLa HSP es una urgencia neurológica real y potencialmente letal. Su diagnóstico es clínico, para lo cual es importante su detección y la educación del personal involucrado en la atención del paciente neurocrítico. El mayor desafío en la práctica diaria actual es realizar una identificación temprana de la HSP, para lo cual es necesario, a corto plazo, homogeneizar y validar tanto la nomenclatura como los criterios diagnósticos, lo que permitirá comparar la eficacia de futuros tratamientos. Resulta imprescindible categorizar precozmente (no todos los paroxismos comparten los mismos rasgos clínicos ni sus características) y trabajar de manera sistemática y multidisciplinaria. Resulta imperioso protocolizar la terapéutica, la cual debería escalarse en función del cuadro clínico exclusivamente. La meta es lograr un tratamiento precoz e intensivo, para prevenir complicaciones, optimizando al máximo las posibilidades de rehabilitación.
Conflicto de interesesNinguno.