


Genomics have allowed important advances in the knowledge of the etiology and pathogenesis of complex disease entities such as acute lung injury (ALI) and acute respiratory distress syndrome (ARDS).
Genomic medicine aims to personalize and optimize diagnosis, prognosis and treatment by determining the influence of genetic polymorphisms in specific diseases.
The scientific community must cope with the important challenge of securing rapid transfer of knowledge to clinical practice, in order to prevent patients from becoming exposed to unnecessary risks.
In the present article, we describe the main concepts of genomic medicine pertaining to ALI/ARDS, and its currently recognized clinical applications.
Recientemente la genómica ha adquirido una enorme relevancia, permitiendo sustanciales avances en el conocimiento de la etiología y patogenia de entidades complejas como la lesión pulmonar aguda (LPA) y el síndrome de distrés respiratorio agudo (SDRA).
La medicina genómica procura personalizar y optimizar el diagnóstico, pronóstico y tratamiento mediante el reconocimiento de la influencia que ejercen los polimorfismos genéticos en enfermedades específicas.
Uno de los principales desafíos que la comunidad científica debe afrontar es lograr que este conocimiento sea transferido pertinente y rápidamente a la práctica clínica. En caso contrario, es posible que los pacientes sean sometidos a un riesgo innecesario.
En el presente artículo se describen los principales aspectos de la medicina genómica en la LPA/SDRA y cuáles son las aplicaciones clínicas actuales.
Approximately 10 years ago, leaders in the United States and the United Kingdom, accompanied by representatives from different scientific groups, announced the first draft of the Human Genome Project (HGP).1 Its publication marked the start of the postgenomic era and radically and irreversibly changed our way of understanding health and disease.2,3 One of the main areas to benefit from this explosion in knowledge was basic medical research, and particularly genomics.4
The 20th century allowed the definition of most genetic diseases, which characteristically involve a single mutation and account for very few deaths, since their frequency in the general population is relatively low. In recent years the concept of disease genetics has been incorporated in reference to the study of the influence of certain nucleotide variants upon the susceptibility to and prognosis of complex and frequent disorders such as, for example, diabetes,5,6 arterial hypertension,7,8 acute lung injury (ALI) and acute respiratory distress syndrome (ARDS).9,10 In contrast to what is seen in genetic diseases, these disorders are characterized by the implication of many genes with normal variants known as polymorphisms. These polymorphisms, when acting in specific settings or contexts, are able to modify patient susceptibility to certain illnesses and/or determine the severity of such illnesses.11
William Osler (1849–1919) was the first to recognize that “variability is the rule in life; in the same way as there are no two identical faces, there are no two identical bodies or two individuals that react in the same way under abnormal circumstances such as disease”.12 Genomic medicine aims to personalize and optimize the diagnosis, prognosis and treatment of diseases through identification of the influence exerted by the normal and frequent genomic variants found among different individuals.
As a result of the extremely rapid growth of genomic or personalized medicine, we have been able to associate hundreds of genetic variants to patient susceptibility to certain diseases, and to the severity of such disorders. Close to 10% of all drugs authorized by the United States Food and Drug Administration (FDA) present pharmacogenomic information on their labeling,13 and use is beginning to be made of diagnostic tests based on the identification of these genetic variants or of other molecular mechanisms capable of individually predicting patient response to certain therapies.14
One of the main challenges facing the scientific community is to ensure that such knowledge is appropriately and rapidly transferred to the clinical setting. Failure to do so may expose some patients to needless risks.
The present article describes the main aspects of genomic medicine in ALI/ARDS, and the clinical applications that are currently available.
Genetic variability: polymorphismsDescription of the human genome revealed the existence of only about 20,000 or 25,000 genes instead of the previously estimated 100,000. A full 99% of the nucleotide sequence is identical from one individual to another2,3; despite this fact, however, each of the approximately 6800 million people on the planet are different and unique. Referred exclusively to the genetic setting, a large proportion of these differences are attributable to the presence of polymorphisms.
Polymorphisms represent normal variants of the deoxyribonucleic acid (DNA) sequence. By definition, a polymorphism is regarded as a variant that is found in over 1% of the population, and which determines the existence of at least two alleles.15 The most frequent of these alleles is referred to as the native or “wild type”, while the other (or others) are referred to as the polymorphic allele or alleles. Since the frequency of the polymorphisms of each gene varies according to the population and geographic setting involved, the different wild type and polymorphic alleles are specific of each population.
In contrast to mutations, polymorphisms considered individually do not cause specific diseases. For example, ALI and ARDS are of course caused by environmental factors such as sepsis or trauma, and there is no single gene responsible for their development. However, variability in terms of susceptibility and/or severity of the disease among individuals is indeed influenced by genetic factors.
Polymorphisms can be of different types:
- •
Single nucleotide polymorphism (SNP), involving the replacement of a single nucleotide by some other nucleotide. Over 3.1 million SNPs have been registered in the human genome,16,17 and as such these are the most frequent type of polymorphism.
- •
Restriction fragment length polymorphism (RFLP), involving specific nucleotide sequences that can be recognized and spliced by restriction enzymes. An individual may or may not have the sequence, and therefore may or may not be polymorphic.
- •
Variable number tandem repeat (VNTR), involving specific and repetitive DNA sequences; the allele exhibiting the most frequent number of repetitions in the population is the wild type, and the rest are polymorphic.18
In turn, polymorphisms can act as follows:
- 1.
Directly, when the presence of the polymorphism is associated to variation in the risk of a given event, for example, mortality or susceptibility versus healthy controls or patients at risk who do not develop the disease.
- 2.
In “linked groups” conforming haplotypes. For example, urokinase is a serine protease that transforms plasminogen into plasmin.19,20 Arcaroli et al.21 reported that the haplotype comprising SNP rs1916341C/rs2227562G/rs2227564C/rs2227566C/rs2227568C/rs4066C is associated to increased mortality at 60 days in patients with ALI and ARDS, despite the fact that each variant considered individually failed to reach statistical significance.
- 3.
In “non-linked groups” conforming combinations of genotypes. As an example, Schroeder et al.22 analyzed the relationship between SNPs of interleukins and susceptibility to ALI/ARDS. They found the presence in one same individual of the genotypes rs114634TC and rs1800872AC to be associated to a 12-fold greater risk of developing ALI, despite the fact that individually these genotypes are not associated to the development of ALI, and that their genes (IL1ß and IL10, respectively) are located on different chromosomes.
One of the main limitations of genetic association studies in ALI/ARDS is the absence of a phenotype that is easily recognizable, objective and reproducible among different observers.
The standard for the diagnosis of ALI/ARDS is the histological confirmation of diffuse alveolar damage. However, such information is generally not available in clinical practice.23,24
The American–European Consensus Conference defined ALI/ARDS on the basis of clinical, radiological and blood gas criteria.25 Many studies have shown that compliance with these criteria depends on the ventilation regimen employed, that these criteria are difficult to recognize, and that there is scant inter-observer agreement in their identification.
Villar et al.26, after applying the same ventilation protocols to 170 patients meeting the consensus criteria, found that the mentioned definition overestimated the incidence of ARDS and underestimated mortality.
The changes in blood gas criterion according to the fraction of inspired oxygen (FiO2) were studied by Gowda and Klocke27 and Ferguson et al.28 Both groups coincided that the PaO2/FiO2 ratio is modified by changes in FiO2. The application of positive-end expiratory pressure (PEEP) and recruitment measures in turn improves oxygenation,29,30 and prone decubitus lessens mortality in extreme situations.31,32 All three procedures are commonly used in the Intensive Care Unit (ICU); however, they were not considered in the definition of the consensus conference.
The radiological pattern was evaluated in two studies–both showing moderate agreement in X-ray interpretation between different observers.33,34
The recognition of ALI/ARDS is extremely difficult even for trained specialists. Ferguson et al.35 studied the case histories of 138 patients subjected to necropsy. They found that only 20 of the 42 patients with histologically confirmed ALI/ARDS had been clinically identified before the time of death.
Lastly, mention should be made of the moderate agreement between the presence of the histological changes and the criteria of the American–European Consensus Conference. Esteban et al.36, in 382 necropsy studies of patients who died in the ICU, found the sensitivity and specificity of the consensus conference definition to be 75% and 84% respectively, in comparison with the histological findings.
Approaches to the genetic study of ALI/ARDSThere are two “classical” approaches to the genetic study of complex diseases: (a) the so-called “genome wide approach” (GWA), and (b) the “candidate gene approach” (CGA). The GWA involves the analysis of a large number of polymorphisms distributed throughout the genome, based on the use of DNA arrays. By knowing a position (locus), structuring of the genome into “haplotypes” makes it possible to predict the adjacent loci to an approximate distance of 3×104 base pairs.37 Therefore, with 50×104 “marker” loci we can study the entire human genome, which contains 3×109 nucleotides.38
The limitations of the GWA are related to the fact that it affords information on loci, but not on genes as such, and interpretation difficulties are found when there are multiple alleles within one same population.39 Studies of this kind attempt to identify which loci are associated to a certain disease, and require the use of complementary techniques such as qualitative polymerase chain reaction (PCR) or DNA sequencing to determine which specific genetic variation is involved.
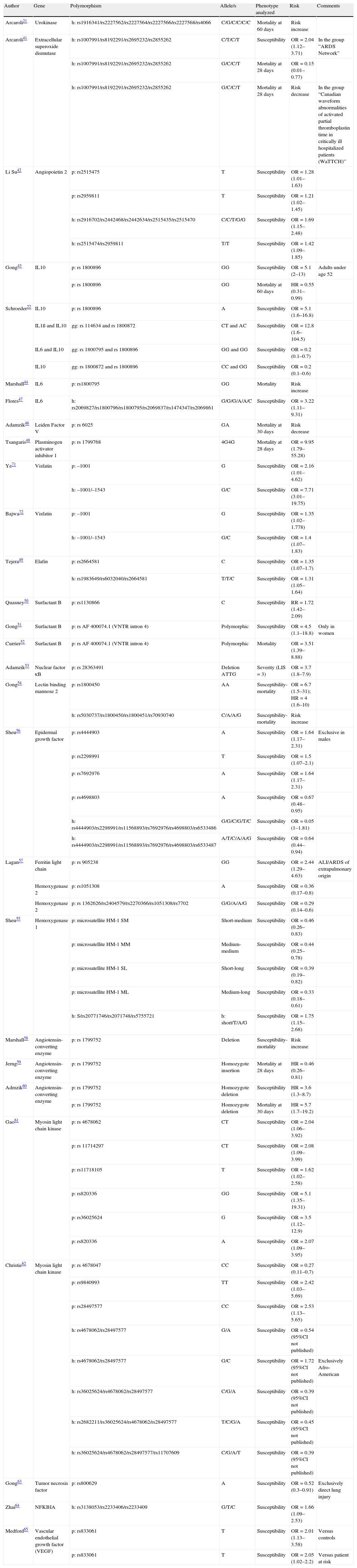
The CGA approach in turn implies the a priori selection of genes and polymorphisms that might be related to the etiopathogenesis and/or physiopathology of the disease.40 The advantages of this approach are its lesser cost and greater technical simplicity—allowing the study of a greater number of individuals while at the same time offering greater solidity of the findings. This technique has been used to identify polymorphisms in 23 genes linked to ALI/ARDS susceptibility and/or mortality (Table 1).41–65 These genetic variants may be of a protective or risk-related nature, with odds ratios (ORs) that range from 0.27 (rs4678047) to 9.95 (rs1799768) when analyzed individually.
Polymorphisms associated to acute lung injury and acute respiratory distress syndrome.
| Author | Gene | Polymorphism | Allele/s | Phenotype analyzed | Risk | Comments |
| Arcaroli21 | Urokinase | h: rs1916341/rs2227562/rs2227564/rs2227566/rs2227568/rs4066 | C/G/C/C/C/C | Mortality at 60 days | Risk increase | |
| Arcaroli41 | Extracellular superoxide dismutase | h: rs1007991/rs8192291/rs2695232/rs2855262 | C/T/C/T | Susceptibility | OR=2.04 (1.12–3.71) | In the group “ARDS Network” |
| h: rs1007991/rs8192291/rs2695232/rs2855262 | G/C/C/T | Mortality at 28 days | OR=0.15 (0.01–0.77) | |||
| h: rs1007991/rs8192291/rs2695232/rs2855262 | G/C/C/T | Mortality at 28 days | Risk decrease | In the group “Canadian waveform abnormalities of activated partial thromboplastin time in critically ill hospitalized patients (WaTTCH)” | ||
| Li Su42 | Angiopoietin 2 | p: rs2515475 | T | Susceptibility | OR=1.28 (1.01–1.63) | |
| p: rs2959811 | T | Susceptibility | OR=1.21 (1.02–1.45) | |||
| h: rs2916702/rs2442468/rs2442634/rs2515435/rs2515470 | C/C/T/G/G | Susceptibility | OR=1.69 (1.15–2.48) | |||
| h: rs2515474/rs2959811 | T/T | Susceptibility | OR=1.42 (1.09–1.85) | |||
| Gong43 | IL10 | p: rs 1800896 | GG | Susceptibility | OR=5.1 (2–13) | Adults under age 52 |
| p: rs 1800896 | GG | Mortality at 60 days | HR=0.55 (0.31–0.99) | |||
| Schroeder22 | IL10 | p: rs 1800896 | A | Susceptibility | OR=5.1 (1.6–16.8) | |
| IL1ß and IL10 | gg: rs 114634 and rs 1800872 | CT and AC | Susceptibility | OR=12.8 (1.6–104.5) | ||
| IL6 and IL10 | gg: rs 1800795 and rs 1800896 | GG and GG | Susceptibility | OR=0.2 (0.1–0.7) | ||
| IL10 | gg: rs 1800872 and rs 1800896 | CC and GG | Susceptibility | OR=0.2 (0.1–0.6) | ||
| Marshall44 | IL6 | p: rs1800795 | GG | Mortality | Risk increase | |
| Flores47 | IL6 | h: rs2069827/rs1800796/rs1800795/rs2069837/rs1474347/rs2069861 | G/G/G/A/A/C | Susceptibility | OR=3.22 (1.11–9.31) | |
| Adamzik46 | Leiden Factor V | p: rs 6025 | GA | Mortality at 30 days | Risk decrease | |
| Tsangaris48 | Plasminogen activator inhibitor 1 | p: rs 1799768 | 4G4G | Mortality at 28 days | OR=9.95 (1.79–55.28) | |
| Ye71 | Visfatin | p: –1001 | G | Susceptibility | OR=2.16 (1.01–4.62) | |
| h: –1001/–1543 | G/C | Susceptibility | OR=7.71 (3.01–19.75) | |||
| Bajwa72 | Visfatin | p: –1001 | G | Susceptibility | OR=1.35 (1.02–1.778) | |
| h: –1001/–1543 | G/C | Susceptibility | OR=1.4 (1.07–1.83) | |||
| Tejera49 | Elafin | p: rs2664581 | C | Susceptibility | OR=1.35 (1.07–1.7) | |
| h: rs1983649/rs6032040/rs2664581 | T/T/C | Susceptibility | OR=1.31 (1.05–1.64) | |||
| Quasney50 | Surfactant B | p: rs1130866 | C | Susceptibility | RR=1.72 (1.42–2.09) | |
| Gong51 | Surfactant B | p: rs AF 400074.1 (VNTR intron 4) | Polymorphic | Susceptibility | OR=4.5 (1.1–18.8) | Only in women |
| Currier52 | Surfactant B | p: rs AF 400074.1 (VNTR intron 4) | Polymorphic | Mortality | OR=3.51 (1.39–8.88) | |
| Adamzik53 | Nuclear factor κB | p: rs 28363491 | Deletion ATTG | Severity (LIS=3) | OR=3.7 (1.8–7.9) | |
| Gong54 | Lectin binding mannose 2 | p: rs1800450 | AA | Susceptibility-mortality | OR=6.7 (1.5–31); HR=4 (1.6–10) | |
| h: rs5030737/rs1800450/rs1800451/rs70930740 | C/A/A/G | Susceptibility-mortality | Risk increase | |||
| Sheu56 | Epidermal growth factor | p: rs4444903 | A | Susceptibility | OR=1.64 (1.17–2.31) | Exclusive in males |
| p: rs2298991 | T | Susceptibility | OR=1.5 (1.07–2.1) | |||
| p: rs7692976 | A | Susceptibility | OR=1.64 (1.17–2.31) | |||
| p: rs4698803 | A | Susceptibility | OR=0.67 (0.48–0.95) | |||
| h: rs4444903/rs2298991/rs11568893/rs7692976/rs4698803/rs6533486 | G/G/C/G/T/C | Susceptibility | OR=0.05 (1–1.81) | |||
| h: rs4444903/rs2298991/rs11568893/rs7692976/rs4698803/rs6533487 | A/T/C/A/A/G | Susceptibility | OR=0.64 (0.44–0.94) | |||
| Lagan57 | Ferritin light chain | p: rs 905238 | GG | Susceptibility | OR=2.44 (1.29–4.63) | ALI/ARDS of extrapulmonary origin |
| Hemoxygenase 2 | p: rs1051308 | A | Susceptibility | OR=0.36 (0.17–0.8) | ||
| Hemoxygenase 2 | p: rs 1362626/rs2404579/rs2270366/rs1051308/rs7702 | G/G/A/A/G | Susceptibility | OR=0.29 (0.14–0.6) | ||
| Sheu55 | Hemoxygenase 1 | p: microsatellite HM-1 SM | Short-medium | Susceptibility | OR=0.46 (0.26–0.83) | |
| p: microsatellite HM-1 MM | Medium-medium | Susceptibility | OR=0.44 (0.25–0.78) | |||
| p: microsatellite HM-1 SL | Short-long | Susceptibility | OR=0.39 (0.19–0.82) | |||
| p: microsatellite HM-1 ML | Medium-long | Susceptibility | OR=0.33 (0.18–0.61) | |||
| h: S/rs20771746/rs2071748/rs5755721 | h: short/T/A/G | Susceptibility | OR=1.75 (1.15–2.68) | |||
| Marshall58 | Angiotensin-converting enzyme | p: rs 1799752 | Deletion | Susceptibility-mortality | Risk increase | |
| Jerng59 | Angiotensin-converting enzyme | p: rs 1799752 | Homozygote insertion | Mortality at 28 days | HR=0.46 (0.26–0.81) | |
| Admzik60 | Angiotensin-converting enzyme | p: rs 1799752 | Homozygote deletion | Susceptibility | HR=3.6 (1.3–8.7) | |
| p: rs 1799752 | Homozygote deletion | Mortality at 30 days | HR=5.7 (1.7–19.2) | |||
| Gao61 | Myosin light chain kinase | p: rs 4678062 | CT | Susceptibility | OR=2.04 (1.06–3.92) | |
| p: rs 11714297 | CT | Susceptibility | OR=2.08 (1.09–3.99) | |||
| p: rs11718105 | T | Susceptibility | OR=1.62 (1.02–2.58) | |||
| p: rs820336 | GG | Susceptibility | OR=5.1 (1.35–19.31) | |||
| p: rs36025624 | G | Susceptibility | OR=3.5 (1.12–12.9) | |||
| p: rs820336 | A | Susceptibility | OR=2.07 (1.09–3.95) | |||
| Christie62 | Myosin light chain kinase | p: rs 4678047 | CC | Susceptibility | OR=0.27 (0.11–0.7) | |
| p: rs9840993 | TT | Susceptibility | OR=2.42 (1.03–5.69) | |||
| p: rs28497577 | CC | Susceptibility | OR=2.53 (1.13–5.65) | |||
| h: rs4678062/rs28497577 | G/A | Susceptibility | OR=0.54 (95%CI not published) | |||
| h: rs4678062/rs28497577 | G/C | Susceptibility | OR=1.72 (95%CI not published) | Exclusively Afro-American | ||
| h: rs36025624/rs4678062/rs28497577 | C/G/A | Susceptibility | OR=0.39 (95%CI not published) | |||
| h: rs2682211/rs36025624/rs4678062/rs28497577 | T/C/G/A | Susceptibility | OR=0.45 (95%CI not published) | |||
| h: rs36025624/rs4678062/rs28497577/rs11707609 | C/G/A/T | Susceptibility | OR=0.39 (95%CI not published) | |||
| Gong63 | Tumor necrosis factor | p: rs800629 | A | Susceptibility | OR=0.52 (0.3–0.91) | Exclusively direct lung injury |
| Zhai64 | NFKBIA | h: rs3138053/rs2233406/rs2233409 | G/T/C | Susceptibility | OR=1.66 (1.09–2.53) | |
| Medford65 | Vascular endothelial growth factor (VEGF) | p: rs833061 | T | Susceptibility | OR=2.01 (1.13–3.58) | Versus controls |
| p: rs833061 | T | Susceptibility | OR=2.05 (1.02–2.2) | Versus patient at risk |
gg, genic group; h, haplotype; HR, hazard ratio; CI, confidence interval; LIS, lung injury score; OR, odds ratio; p, polymorphisms; rs, reference sequence.
The studies found in the literature analyze a limited number of genes. Given the undeniable interaction among genes, and therefore among their polymorphisms, it would be important to conduct studies to explore different variants in one same setting. As an example, an association has been demonstrated between ALI/ARDS with genes encoding for acute phase reactant producers (MBL2), cytokines (IL10, IL1ß, IL6, TNFα) and immune response regulators (NKBIA). However, they have not all been jointly analyzed in one same cohort. Polymorphisms exert specific effects in certain subgroups. As an example, Lagan et al.57 demonstrated that the rs905238 GG genotype of the ferritin light chain is a risk factor for ALI/ARDS exclusively when the causal disease is of extrapulmonary origin. Gong et al.43 in turn found that the SNP IL10 rs1800896 (+1082) GG increases susceptibility in patients under 52 years of age, and Sheu et al.56 published similar results for rs444490 (+61) A of the gene encoding for epidermal growth factor, exclusively in males.
Some investigators are currently attempting integral approaches, combining different molecular biological techniques. An interesting example is the case of the gene encoding for visfatin (a pre-B cell colony-enhancing factor, PBEF). This molecule is an adipokine related to the generation of pre-B cell colonies, normal and preterm delivery, sepsis, colorectal cancer, obesity and diabetes.66–70 Ye et al.,71 using expression arrays, analyzed the genic products in respiratory cells obtained from bronchoalveolar lavage (BAL) in patients with ALI/ARDS and healthy controls, and in lung tissue obtained from canine and murine models of ALI. The authors found visfatin expression to be significantly increased in the three species (3.79-fold in humans 5.79-fold in dogs, and 2.13-fold in rats). Posteriorly, they sequenced the entire gene in some patients with ALI/ARDS, with sepsis, and in healthy controls, with the purpose of identifying the possible SNPs. The authors found SNP-1001T/G to be over-expressed in the group of patients with ALI/ARDS versus the other two groups. Subsequently, genotyping was carried out in 87 individuals with ALI/ARDS, in 100 patients with sepsis and in 84 healthy controls. It was thus shown that the “G” allele increased the risk of developing ALI/ARDS 2.16-fold. Similar results were reported by Bajwa et al.72, who studied SNPs-1001T/G and -1543C/T of visfatin in 375 patients with ALI/ARDS and in 787 critical patients.
Validity of the resultsGenetic association studies have increased exponentially in recent years. The two main types of genetic association studies correspond to cohort studies and case–control series. Beyond the statistical analysis used, some fundamental aspects must be taken into account in order for the results to be valid with both types of study design73:
- •
A clear and specific definition of the disease and its phenotype. As has been commented above, this aspect represents one of the major limitations of ALI/ARDS studies.
- •
A clear and specific definition of the population. Genes act in concrete contexts. As a result, it is essential to correctly characterize the study population, its origins, racial composition, etc. Genetic results obtained on a blind basis with respect to the clinical information.
- •
Control of genotyping errors. Such errors can vary from less than 1% to as much as 30%.74 They can occur at any time from obtainment of the biological sample to reading of the results. It is necessary to clearly define the chain of events experienced by the biological material, and the strategies used for error control.
- •
Hardy–Weinberg equilibrium (HWE). This principle states that under certain conditions, the genotypes of a population remain stable. In the simplest case of a locus with two alleles A and a, the allelic frequencies of which are p and q, respectively, the HWE principle predicts that the genotypic frequency for the homozygote AA is p2, versus 2pq for the heterozygote and q2 for the homozygote aa. Due consideration of the HWE is essential, since its absence may be linked to population or laboratory factors capable of invalidating the results obtained (Table 2).75,76 In case–control studies, the equilibrium must be sought in the control group, while in the case of cohort studies the equilibrium is normally analyzed in the global sample.
- •
Adjusting significance for multiple comparisons. This correction, applicable both to the HWE and to the genotyping results when multiple variants are examined in the same sample, is one of the most frequent causes of false positive associations. The simplest correction is the Bonferroni correction, which involves dividing the P-value by the number of associations made to obtain significance. As an example, if we consider a P-value of 0.05 and 15 tests are made, the real significance will be 0.05/15, i.e., 0.003.
Reporting or communication of the results must be clear and concise, offering the reader and reviewer the information needed to interpret the findings.
Flores et al.77 analyzed the quality of the genetic association studies. The measure of quality, using a scale from 0 to 10 points, was 6.62 points with an interval of 0.71–7.4. On considering the studies according to their year of publication, a tendency towards improvement over time was noted–particularly in reference to the case–control studies.
In an effort to improve reporting, and adopting the general guidelines of the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE),78 the so-called recommendations for Strengthening the Reporting of Genetic Association Studies (STREGA) were published in 2009.79 The recommendations of the STREGA are organized in the form of 5 main groups: genotyping errors, population stratification, haplotype models, Hardy–Weinberg equilibrium, and replication of the results. The adopted format (check list) makes it possible to homogenize the reports, and facilitates the work of the reviewers of the different journals.
From the laboratory to the clinical settingIt is practically universally accepted among clinicians that most complex diseases are influenced by the genic structure of the individual. However, it is still widely and subjectively believed that such genomic knowledge lacks practical applications.
The truth is that genomic medicine is quickly and continuously expanding its influence in daily clinical practice. At present, its main applications can be summarized as follows:
- •
Risk identification and quantification. This presently represents the main application of genomic medicine. Genetic risk factors, in contrast to the traditional risk factors, are present and can be identified at any point in life—thus offering a unique opportunity for the adoption of preventive measures. As an example, it has been shown that the risk of dying of an infection is familial and inheritable.80 Consequently, it could be important to identify genetic variants associated to sepsis/septic shock in blood relatives of patients who have suffered such a disease.
- •
Optimization of the diagnosis. The availability of a “genomic signature” in ALI/ARDS could help solve the diagnostic difficulties associated with this syndrome.
- •
Generation of new etiological and physiopathological knowledge. Modern medicine favors the development of innovating hypotheses and lines of research. The identification of genic variants will help orientate the development of knowledge towards specific goals, while also contributing to the development of innovating and individualized treatments.
- •
Pharmacogenetics is the science that studies how genetic differences condition patient respond to drug treatment. It facilitates the prescription of drugs in specific patients with the purpose of deriving increased benefits and of minimizing the adverse effects.81,82
- •
Nutrition genomics is the science that studies the expression of genes in relation to nutrition and the development of diseases associated to such expression. This field of medicine helps our understanding of the interaction between the environment and genes.
The challenges facing genomic medicine in the coming years include83: (a) the replication of studies by independent groups, since confidence increases exponentially when the results are effectively reproduced by different groups involving independent samples; (b) the study of populations involving a larger sample size, with the purpose of reducing the incidence of false positive and negative associations; (c) extension of research to different populations, striving to identify new genetic variants and generalize the results; (d) the study of “rare” variants, for while there are frequent variants with a limited individual influence, there also may be rare variants with an important influence; and (e) expansion of our knowledge of the influence of genic variants upon disease pathogenesis.84
The future of genomic medicine is fundamentally based on the very rapid development of molecular techniques, particularly in reference to DNA sequencing. The first generation (chain-termination method) in DNA sequencing was described by Sanger et al.,85,86 in the late 1970s. Posteriorly, in the year 2005, the second generation (wash-and-scan) was developed with the purpose of reducing costs and boosting production. This technology is based on the anchoring of tens of thousands of identical chains in specific positions to be identified in a wash-and-scan process. The DNA anchoring matrix may have a great density of DNA fragments, giving rise to an extremely high overall yield at a cost per identified nucleotide far lower than that of the traditional method. In recent years, the third sequencing generation (SMS, single-molecule sequencing) has been developed as an option for further increasing the yield at a much lesser cost (both economical and in terms of time). This latter generation comprises a group of techniques mainly characterized by non-interruption of the sequencing process after incorporation of a nucleotide.87
With the introduction of techniques capable of segregating the exon sequences88–90 from the rest of the DNA, the possibility of constructing “complete exomes” has become a reality. The study of exomes, with a size of only 30Mb, is particularly important in monogenic diseases, since most of the genic variants determining them are located in exons or in splicing sites. Although ALI/ARDS is not monogenic, exploring the exome could be very useful, since if monogenic traits are identified, specific subgroups could be defined, thus orientating research towards new “candidate genes”.
ConclusionsPublication of the human genome afforded the context and the impulse needed for the development of genomic or “personalized” medicine.
Genomic medicine aims to personalize and optimize the diagnosis, prognosis and treatment of diseases, based on identification of the influence exerted by normal and frequent genomic variations among different individuals.
ALI/ARDS is a serious, complex and multigenic disease with a “clinical” phenotype as defined by the American–European Consensus Conference that poses serious limitations.
At present, practically all studies published on the genetics of ALI/ARDS make use of the analysis of candidate genes. Recently, emphasis has been placed on the difficulties inherent to the design of the studies, the validity of the findings and the way in which they are reported; as a result, general criteria have been developed that should be considered when interpreting the data obtained and preparing the manuscripts.
The future of genomic medicine centers on the development and optimization of the molecular biological techniques that will allow us to ensure the massive production of information at a lesser cost—thereby making it possible to reach the established goals.
Conflicts of interestThe authors have no conflicts of interest to declare.
Please cite this article as: Cardinal-Fernández P, et al. Lesión pulmonar aguda y síndrome de distrés respiratorio agudo: una perspectiva genómica. Med Intensiva. 2011;35:361–72.

