



In the neurocritical care setting, hyponatremia is the commonest electrolyte disorder, which is associated with significant morbimortality. Cerebral salt wasting and syndrome of inappropriate antidiuretic hormone have been classically described as the 2 most frequent entities responsible of hyponatremia in neurocritical care patients. Nevertheless, to distinguish between both syndromes is usually difficult and useless as volume status is difficult to be determined, underlying pathophysiological mechanisms are still not fully understood, fluid restriction is usually contraindicated in these patients, and the first option in the therapeutic strategy is always the same: 3% hypertonic saline solution. Therefore, we definitively agree with the current concept of “cerebral salt wasting”, which means that whatever is the etiology of hyponatremia, initially in neurocritical care patients the treatment will be the same: hypertonic saline solution.
En el paciente neurocrítico la hiponatremia es la distonía más frecuente, comportándose como un predictor pronóstico. Clásicamente, el cerebro perdedor de sal y la secreción inadecuada de hormona antidiurética han sido las 2 entidades responsables de explicar la mayor parte de los casos de hiponatremia en estos pacientes. Sin embargo, en virtud de la dificultad en establecer el estado de la volemia en el paciente crítico, el diagnóstico diferencial es con frecuencia difícil de establecer. Por otra parte, en el paciente neurocrítico el diagnóstico diferencial entre ambos síndromes no ha demostrado ser de utilidad debido a que el cloruro de sodio hipertónico es la piedra angular en el tratamiento de ambos cuadros, y la restricción hídrica con frecuencia está contraindicada. Es por ello que ha surgido el concepto de «cerebro falto de sal», lo cual traduce la necesidad del aporte de sodio como estrategia terapéutica en todos los casos.
Hyponatremia is defined as a serum sodium concentration of <136mmol/l, and is the most common electrolyte disorder in hospitalized patients (affecting 15–20% of all individuals requiring hospital admission).1,2 Likewise, hyponatremia is a mortality predictor in critical patients. In this regard, Stelfox et al.3 have shown that hyponatremia acquired in the Intensive Care Unit (ICU) increases hospital mortality by 16–28% (p<0.001). More recently, Sturdik et al.4 have demonstrated that an age of over 65 years, the presence of dilutional hyponatremia, and its inadequate correction are three independent risk factors associated to increased hospital mortality in hyponatremic patients. Corona et al.,5 in a systematic literature review and metaanalysis including 80 randomized clinical trials (RCTs), showed hyponatremia–even when mild–to be associated to a significant increase in mortality in the ICU (RR: 2.60; 95%CI: 2.31–2.93, p<0.0001).5 Likewise, inadequate treatment of this electrolyte disorder, failing to observe the required correction range (overcorrection), as well as insufficient treatment, imply added risk that further worsens the prognosis of critical patients with hyponatremia.6,7
In neurocritical patients, hyponatremia is also the most common electrolyte disorder, having been reported in up to 50% of all cases of serious neurological injury.8 Among the acute brain conditions, severe traumatic brain injury (TBI) and aneurysmal subarachnoid hemorrhage (SAH) are those with the highest incidence of hyponatremia. In this regard, Sherlock et al.9 showed hyponatremia to be more frequent in patients with hypophyseal disease (5/81; 6.25%), TBI (44/457; 9.6%) and intracranial tumors (56/355; 15.8%).9 In their study the authors found that among 316 patients with SAH, 179 (56.6%) developed hyponatremia, which proved severe (natremia <130mmol/l) in 62 cases (19.6%). In SAH, hyponatremia can be due to different causes, including syndrome of inappropriate antidiuretic hormone secretion (SIADH), so-called cerebral salt wasting (CSW)–also known as renal salt wasting–and pressure natriuresis or acute corticosteroid deficiency (hypocortisolism).10–13 Up until now, CSW has been regarded as the most frequent cause of hyponatremia in the evolutive course of aneurysmal SAH, though recently this has been strongly questioned. However, on the basis of current knowledge, the differential diagnosis between SIADH and CSW is often difficult to establish, and both syndromes moreover have been suggested to be part of one same disease condition–manifesting successively in the same patient.10,11
Considering the above, the present review was carried out to provide an update on the etiological and physiopathological aspects of hyponatremia in the neurocritical patient, placing special emphasis on the treatment strategies adjusted to current evidence.
Hypoosmolarity and neuronal adaptation mechanismsHyponatremia/hypoosmolarity is a primary cause of water entry to the cells, resulting in an increase in cell volume (volumetric variation secondary to osmotic change).14 This change in turn triggers a volume-regulating mechanism called regulatory volume decrease,15 characterized by a rapid potassium, chloride and sodium outflow phase with the purpose of quickly “buffering” the osmotic change, and a second outflow phase of organic osmolytes (“non-perturbing” osmolytes) which accumulate in the neurons without producing deleterious effects upon cell structure and function (cytoprotective function). These compensating mechanisms are incomplete in acute hyponatremia (evolution under 48h) and complete in chronic hyponatremia (evolution longer than 48h). In the presence of cell swelling, a first phase (called the rapid phase) is observed, involving osmolyte outflow, followed by a second phase (or slow phase) characterized by inhibition of the synthesis of these osmolytes.16 However, in acute neurological injury these protective mechanisms carried out by the neuroglia in response to plasma hypotonicity are altered. Likewise, the increase in circulating antidiuretic hormone (ADH) levels observed in the two characteristic conditions (CSW and SIADH) results in action upon the V1a receptors of vascular smooth muscle. The stimulation of these receptors generates vasoconstriction independent of the endothelium, due to an increase in the calcium levels through activation of the phosphatidylinositol-bisphosphonate cascade.17 This cerebral vasoconstrictor effect reduces cerebral blood flow, oxygen supply to the astroglia, and the production of ATP and phosphocreatine.17
Etiology and physiopathologyCerebral salt wasting and SIADH have been defined as the most common causes of hyponatremia in the neurocritical patient.18,19 However, there are other less prevalent etiologies, such as the hypervolemic presentations and pressure natriuresis, which also must be discarded in order to establish an adequate diagnostic strategy.20
Likewise, despite the notorious interest in these two syndromes in recent years, the differential diagnosis between CSW and SIADH is often a genuine challenge, since the clinical manifestations are not always conclusive, and the laboratory test information is frequently confusing. In this regard, the current tendency is to classify both conditions under one same disease entity.16,21
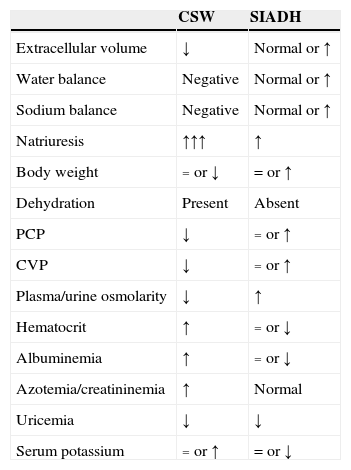
Table 1 shows the main characteristics allowing us to establish a differential diagnosis between CSW and SIADH.2,8,12,16
Conventional schematic representation of cerebral salt wasting (CSW) and syndrome of inappropriate antidiuretic hormone secretion (SIADH).
| CSW | SIADH | |
|---|---|---|
| Extracellular volume | ↓ | Normal or ↑ |
| Water balance | Negative | Normal or ↑ |
| Sodium balance | Negative | Normal or ↑ |
| Natriuresis | ↑↑↑ | ↑ |
| Body weight | = or ↓ | = or ↑ |
| Dehydration | Present | Absent |
| PCP | ↓ | = or ↑ |
| CVP | ↓ | = or ↑ |
| Plasma/urine osmolarity | ↓ | ↑ |
| Hematocrit | ↑ | = or ↓ |
| Albuminemia | ↑ | = or ↓ |
| Azotemia/creatininemia | ↑ | Normal |
| Uricemia | ↓ | ↓ |
| Serum potassium | = or ↑ | = or ↓ |
CSW: cerebral salt wasting; PCP: pulmonary capillary pressure; CVP: central venous pressure; SIADH: inappropriate antidiuretic hormone secretion syndrome; ↑: increased; ↓: decreased; =: without changes.
Classically, the presence of hypovolemia has been regarded as the distinctive feature of CSW, though it is difficult to diagnose in the neurocritical patient.
However, a series of methods are now available for assessing volemia and cardiac output in the critical patient. With the introduction of hemodynamic monitoring devices fundamented upon arterial pulse wave analysis (PiCCO®, PulseCO®, FloTrac/Vigileo® and MostCare®, among others), it has become simple to estimate these parameters on a continuous basis.22 The use of such invasive techniques provides adequate results that are better than static preload values such as central venous pressure or pulmonary artery wedge pressure.23 The use of a device of this kind may be necessary in order to obtain a good evaluation of patient volemia, and therefore an adequate diagnosis of the type of hyponatremia involved. Likewise, a noninvasive technique widely used in the ICU, such as ultrasound, can prove useful for estimating volemia in patients of this kind.
Recently, Gritti et al.24 have evaluated the cumulative sodium balance with the purpose of diagnosing CSW in 35 neurocritical patients. In these individuals, the authors showed that the risk of developing hypovolemia is increased 7.1-fold (p<0.001) in the presence of a negative sodium balance. Likewise, the multivariate analysis identified a negative water balance and a negative sodium balance of >2mEq/kg as independent risk factors for hypovolemia. A negative sodium balance was therefore identified as a useful tool for establishing a differential diagnosis between CSW and SIADH in neurocritical patients.24
Hyponatremia in specific clinical scenariosSubarachnoid hemorrhageHyponatremia is highly prevalent in patients with SAH, being described in 10–50% of the cases, particularly in high-grade SAH, in the presence of anterior circulation aneurysms, and in hydrocephalia. In 2005, Kao et al.25 found 22.9% of the case of hyponatremia to be secondary to CSW, while 35.4% corresponded to SIADH. In an elegant and recent study, Hannon et al.26 prospectively evaluated the etiology of hyponatremia in a sample of 100 patients with SAH, based on clinical assessment and the determination of serum cortisol, arginine, vasopressin and brain natriuretic peptide. In their study,26 the authors identified SIADH as the predominant cause (71.4%), while hypocortisolism accounted for 8.2% of the cases of hyponatremia. The remaining 20.4% of the cases were in turn attributed to hypovolemia or to the type of fluid administered. However, the most significant finding of the mentioned study was the absence of cases consistent with CSW.26
Nakagawa et al.27 have recently demonstrated that the early administration of fludrocortisone acetate can reduce the risk of hyponatremia (26% versus 16%) and of symptomatic vasospasm (18.5% versus 6.1%) in SAH. These data could indicate the existence of an association between hyponatremia and the development of vasospasm, though further studies are needed to clarify this relationship.27 Likewise, in SAH, triple H therapy (hypertension, hypervolemia and hemodilution) as an anti-vasospasm strategy promotes pressure natriuresis and the risk of hyponatremia.
Ischemic strokeThe presence of hyponatremia in ischemic stroke has been proposed as a predictor of poor outcome, though the underlying etiopathogenesis is not fully clear. Huang et al.,28 in 925 patients with a first episode of ischemic stroke, recorded an incidence of hyponatremia of 11.6%, which in turn was associated to a significant increase in mortality after three years (RR: 2.23; 95%CI: 1.30–3.82%). Likewise, Rodrigues et al.29 recently identified the presence of hyponatremia in 16% of 565 stroke patients. In this population, hyponatremia was associated to a significant increase in hospital mortality (p=0.039) and in mortality after three months (p=0.001) and one year (p=0.001). A surprising observation was a greater incidence of urinary infection in the patients with hyponatremia. Lastly, hyponatremia associated to a first episode of ischemic stroke is an independent risk factor for the development of seizures, according to Wang et al.30 (OR: 2.10) and Roivainen et al.31 (OR: 3.26; 95%CI: 1.41–7.57).
Traumatic brain injuryHyponatremia is highly prevalent in patients with severe TBI, where dysfunction of the hypothalamic–hypophyseal–adrenal axis is common,32 having been described in 15–68% of the cases, with an incidence of hypopituitarism of 50%33 in the course of the disorder. In severe TBI, hypopituitarism is more frequent in younger patients, as well as in those administered etomidate, propofol or fenobarbital.32 The clinical manifestations include hypocortisolism, hyponatremia, hypoglycemia and arterial hypotension.34 In clinical practice the systematic assessment of hypophyseal function has been recommended in patients with skull base fractures, diffuse axonal damage, and in cases of prolonged admission to the ICU. However, despite the high incidence of hypocortisolism, the existing evidence contraindicates the empirical use of corticosteroids as specific treatment for intracranial hypertension in TBI.
The analysis of other causes of hyponatremia in TBI shows SIADH to account for 33% of the cases, presenting a particular association to subdural hematoma and focal contusions.32–34 Likewise, SIADH often manifests during the second week of admission to the ICU and during the evolution of central or neurogenic diabetes insipidus–this being explained by the release of ADH stored in the axons of the neurohypophysis.21
Lastly, it must be mentioned that in patients with severe TBI, hyponatremia may be secondary to the use of certain drugs such as 20% mannitol–administered as an osmotically active agent for the control of intracranial pressure (hypovolemic stimulus secondary to osmotic diuresis)–carbamazepine and desmopressin (iatrogenic hyponatremia), used for the treatment of the central diabetes insipidus.32–34
NeurosurgeryIn a retrospective analysis including 291 children subjected to tumor resection published by Hardesty et al.,35 hyponatremia was present in almost 10% of the patients, and 5% met the previously established criteria of CSW.
Trans-sphenoidal surgery is one of the surgical procedures most commonly associated to hyponatremia, manifesting between 5 and 9 days after the operation, with a peak incidence on day 7. Such hyponatremia may be early (secondary to axonal degeneration and the massive release of ADH) or late (often secondary to adrenal gland insufficiency).21 Jahangiri et al.,36 in 1045 consecutive surgeries, recently recorded an incidence of hyponatremia of 3% in the preoperative period–a figure that increased to 16% in the postoperative period. In turn, 19% of the hyponatremia episodes were asymptomatic; the mean timing of appearance was day 4; and preoperative hypopituitarism was the only variable associated to an increased risk of postoperative hyponatremia. Likewise, hyponatremia was present in 15% of the cases requiring hospital admission. Lastly, the authors concluded that SIADH was the predominant syndrome in this population–CSW being exceptional.36 In turn, Hussain et al.,37 in 339 neurosurgical patients, recorded a 15% incidence of postoperative hyponatremia in the first 30 days after surgery. Of these episodes, 50% were regarded as mild, and hospital readmission proved necessary in 6.4% of the patients.
Treatment of hyponatremiaIndependently of the underlying etiology and of the fact that the physiopathology involved is not always clear, the presence of hyponatremia in neurocritical patients justifies grouping of the two classical hyponatremia-producing syndromes in one same treatment strategy.
The treatment of hyponatremia recognizes central pontine myelinolysis as the most severe and feared complication, being related to overcorrection, while insufficient replacement may also be a cause of permanent brain damage.38 For this reason, in neurocritical patients, the treatment of hyponatremia requires identification of its severity, duration (acute and chronic forms), patient volemia status, and the severity of the clinical condition. In these patients, and depending on the evolutive stage, the appearance of hyponatremia must be regarded as a genuine medical emergency requiring the immediate adoption of adequate treatment measures. The following sections analyze the management strategies referred to hyponatremia in different clinical scenarios, including hyponatremic encephalopathy and a number of critical neurological disorders.
Severe and symptomatic hyponatremia: hyponatremic encephalopathyRecently, the European Society of Intensive Care Medicine, in coordination with the European Society of Endocrinology and the European Renal Association-European Dialysis and Transplant Association, represented by the European Renal Best Practice, have published good clinical practice guides on the diagnosis and treatment of hyponatremia in critical patients.38 In the case of severe and symptomatic hyponatremia (hyponatremic encephalopathy), these guides recommend the intravenous infusion of 150ml of 3% hypertonic saline solution (HSS) over 20min. This 3% HSS bolus is to be repeated in the next 20min as long as the symptoms persist or if natremia fails to increase significantly. In this regard, dosing can be repeat up to two times or until a natremia increment of 5mmol/l is achieved.38 The infusion of 3% HSS is able to increase natremia by 1–2mmol/h–reversion of the symptoms of hyponatremic encephalopathy (seizures, altered consciousness) requiring an increase of 4–6mmol38 (see Fig. 1).

In a way similar to the recommendations of the European Society of Intensive Care Medicine/European Society of Endocrinology/European Renal Association-European Dialysis and Transplant Association, Sterns et al.,20 in the presence of severe symptoms, recommend the administration of 100ml of 3% HSS via the intravenous route during 10min, with the possibility of repeating the dose three times if needed, in order to control the clinical condition. Although there are more conservative regimens comprising 6mmol/day, it is important to emphasize that the natremia increment when the starting concentration is ≤120mmol/l should not exceed 10mmol/l during the first 24h of 3% HSS administration–with a maximum increase of 8mmol/l in the 24h after obtaining a natremia level of 130mmol/l.12,20,38 Thus, the increase in serum sodium should not exceed 18mmol/l in 48h. Lastly, it must be taken into account that the correction of concomitant hypokalemia also favors the correction of hyponatremia, with the additional need to correct the hypoxemia in order to favor neuronal adaptation to the hypotonic damage.
Treatment of hyponatremia in the neurocritical patientThe hyponatremia management strategy in neurocritical patients implies treatment of the underlying cause in parallel to management of hyponatremia per se. In current clinical practice in neurological ICUs, establishing a differential diagnosis between CSW and SIADH should no longer be seen as a crucial step for guiding treatment.12,20 It has classically been considered that the treatment of CSW requires vigorous sodium administration in the form of 3% HSS, with the purpose of compensating natriuresis. In contrast, SIADH regards fluid restriction as the cornerstone of treatment, since the renal reabsorption of free water is increased in SIADH. However, this strategy is relatively contraindicated in patients with SAH, due to the high risk of vasospasm (level of evidence ii). Likewise, other management measures that can be adopted in SIADH are the use of lithium, demeclocycline and furosemide (Fig. 1).
In 2008, Sterns and Silver12 introduced the concept of cerebral salt wasting, which assumes that in patients with severe brain injury, the presence of hyponatremia requires the administration of 3% HSS independently of the cause underlying the sodium disorder.12
On the other hand, in neurocritical patients with high ADH and natriuretic peptide levels, the administration of isotonic saline solution generates the so-called desalination phenomenon,39 which can worsen hyponatremia secondary to natriuresis, with the renal production of electrolyte-free water.
3% Hypertonic saline solutionTo date, and despite the high prevalence of hyponatremia in neurocritical patients, no randomized clinical trials (RCTs) have evaluated different hypertonic fluids in the treatment of hyponatremia. Likewise, the existing evidence on the use of HSS is based on non-randomized trials; as a result, the posology and duration of treatment cannot be clearly defined.40,41
Three percent HSS can safely increase natremia without causing neurological, cardiac or renal alterations, and is often administered via a peripheral venous access, with a low risk of complications.40,41 This solution contains the minimum sodium concentration capable of exceeding the maximum osmolar concentration of urine. As a result, if 3% HSS constitutes the only replenishment source, natremia will always increase, since the desalination phenomenon is highly improbable.
Current evidence indicates that the continuous infusion of 3% HSS is safe in the presence of strict monitoring of patient natremia, complying with the correction ranges, and when the maximum target natremia is 155–160mmol/l (permissive or therapeutic hypernatremia).42 In this regard, Aiyagari et al.43 reported a 52% mortality rate in severe hypernatremia defined as >160mmol/l (induced or spontaneous), in a population of neurocritical patients. Likewise, Froelich et al.44 showed the appearance of hypernatremia >155mmol/l in neurocritical patients subjected to the continua perfusion of 3% HSS to be associated to the development of renal dysfunction, with creatininemia >1.50mg/dl (p=0.01).
Three percent HSS infusion can cause side effects such as hypernatremia, hyperchloremia, hyperchloremic metabolic acidosis, hypokalemia and acute renal failure45 (Table 2). On the other hand, an initial intravenous bolus may cause transient arterial hypotension, as well as hypervolemia and hemodynamic pulmonary edema. The administration of a solution of sodium lactate 0.5M (osmolarity 1008mOsm/l) with the purpose of reducing the episodes of intracranial hypertension in patients with TBI is able to correct natremia without producing hyperchloremia or metabolic acidosis, as recently demonstrated by Ichai et al.46
Drug intervention in the neurocritical patient with hyponatremia.
| Strategy | Usual dose | Side effects | Indications |
|---|---|---|---|
| 3% Hypertonic saline solution20,38,40–45: sodium 513mmol/l; chloride 513mmol/l; osmolarity 1026mOsm/l | The usual doses for treatment of the symptomatic and asymptomatic forms are shown in Fig. 1 | Overcorrection, thrombosis, phlebitis, hypervolemia, hyperchloremia, acidosis, lung edema | Symptomatic acute or chronic hyponatremia. Osmotherapy in ICH. Osmotic diuresis due to natriuresis and decreasing natremia. Permissive or therapeutic hypernatremia |
| Fludrocortisone38,47–50 | 0.1–0.2mg/8–12h (oral or enteral route) | Hypervolemia, lung edema, hypokalemia | Hypovolemic hyponatremia, CSW |
| Conivaptan38,53,54 | 20mg i.v. in 30min in loading dose, followed by continuous i.v. perfusion of 20–40mg/day. Duration under 4 days | Overcorrection, hypokalemia, orthostatic hypotension, hyperazotemia | Euvolemic and hypervolemic hyponatremia. SIADH (not in moderate to severe forms). Contraindicated in CSW, high risk of vasospasm, glomerular filtration rate <30ml/min or severe liver dysfunction |
| Tolvaptan38,59,60 | 15mg on day 1, possibility of adjusting to 30–60mg with intervals of 24h if natremia is <135mmol/l or the increase is <5mmol/l/d | ||
CSW: cerebral salt wasting; ICH: intracranial hypertension; i.v.: intravenous route; SIADH: syndrome of inappropriate antidiuretic hormone secretion.
In 2005, the first systematic literature review and metaanalysis on the effects of corticosteroids in aneurysmal SAH and primary intracerebral hemorrhage, including 8 RCTs (n=256), evidenced no positive effect with the administration of fludrocortisone.47
Fludrocortisone acetate is a synthetic mineralocorticoid that favors the renal tubular reabsorption of sodium. It is able to reduce the natriuretic response, thereby preventing the appearance of hyponatremia.48 On the other hand, thanks to its weaker glucocorticoid effect, fludrocortisone is associated to a lesser incidence of side effects compared with hydrocortisone. In 2009, a multidisciplinary committee recommended the use of fludrocortisone in patients with SAH who develop hyponatremia and are thus at a high risk of vasospasm. In turn, the Stroke Council of the American Heart Association49 recommends the use of HSS and fludrocortisone as a reasonable strategy for the treatment of hyponatremia and the reduction of effective circulating volume in aneurysmal SAH. Likewise, in October 2010, the consensus conference of the Neurocritical Care Society on the management of aneurysmal SAH established that early treatment with fludrocortisone can be used to limit natriuresis (moderate quality evidence, weak recommendation).50
However, it is necessary to point out that fludrocortisone acetate loses efficacy when hyponatremia is severe. In 2009, Nakagawa et al.,51 in a series of 39 patients with aneurysmal SAH, found high atrial natriuretic peptide values to be associated to hyponatremia. The authors postulated that early treatment of the latter–including the use of fludrocortisone–would be able to inhibit the late ischemic neurological deficit, since the mentioned drug can reduce the incidence of symptomatic vasospasm. In this regard, they suggested that an increase in urine sodium levels is a good indicator for starting enteral fludrocortisone in the early stages of SAH.51 At present, in neurocritical patients with osmotic diuresis and natriuresis (CSW), the recommended daily dose of fludrocortisone is 0.1–0.2mg via the oral route, 2–3 times a day. This treatment is to be maintained until the normalization of natremia and volemia is–this often being achieved after 3–5 days of treatment. Prolonged fludrocortisone treatment increases the incidence of side effects such as hypokalemia, arterial hypertension and hydrostatic pulmonary edema (Table 2). Likewise, mineralocorticoid escape is a well known phenomenon that conditions tolerance to its effects and a loss of action of the drug after administration periods of over one week.
On the other hand, hydrocortisone has been shown to be effective for the control of natriuresis in patients with SAH and hyponatremia,50 though the existing data are scarce, and further RCTs are needed to adequately assess the use of this strategy in neurocritical patients. Lastly, the combined use of hydrocortisone and fludrocortisone has not been shown to exert synergic effects in terms of sodium gain in this group of patients.
Vasopressin antagonists (vaptans)Two drugs of this class, tolvaptan and conivaptan, have been approved to date by the United States Food and Drug Administration (FDA) for the treatment of normovolemic and hypervolemic hyponatremia.52
Conivaptan, administered via the intravenous route, is a receptor V1a and receptor V2 antagonist, and is the drug of choice in the hospital setting for those patients unable to tolerate the oral route. However, its use is limited to a maximum of four days,53 and its effect is greater in the presence of hyponatremia and a high glomerular filtration rate (GFR). Likewise, the administration of conivaptan in intermittent doses has been found to be effective in increasing the renal excretion of free water–with the consequent correction of hyponatremia in neurocritical patients.54 In effect, Murphy et al.54 recorded a minimum increase in natremia of 6mmol/l in up to 52% of the patients, while 69% of them showed a rise in natremia of 4mmol/l after 72h. More recently, Human et al.55 conducted a retrospective evaluation of 124 neurocritical patients, and found the administration of an intravenous bolus dose of conivaptan to be able to produce a mean increase in natremia of 4mmol/l.
On the other hand, conivaptan has been shown to reduce intracranial pressure and increase natremia on a transient basis, without adverse effects, in patients with severe TBI.56 In this sense it reflects the concept of reductive osmotherapy, in which there is no gain in solute but a decrease in solvent. On the other hand, this effect could also be related to inhibition of the V1a receptor, which intervenes in the expression of AQP4 aquaporin channels this being of fundamental importance in the development of brain edema in patients with severe TBI and stroke.57,58 However, this requires confirmation by further studies. The current conivaptan dosage consists of a 20mg intravenous loading dose administered over 30min, followed by a continuous intravenous perfusion of 20–40mg/day for a period of under four days.
Tolvaptan in turn is a receptor V2 antagonist59 that has shown clinical benefits by reducing hospital stay in patients with SIADH, as evidenced by the SALT-1 and SALT-2 trials carried out in North America and Europe, respectively.60 In case reports, this drug has been found to be safe and effective in the treatment of hyponatremia associated to SIADH in severe TBI60 and in patients with non-traumatic neurological damage, where the administration of a daily dose of 15mg of tolvaptan has been seen to be as effective as conivaptan.60 The currently recommended tolvaptan dose is 15mg, which can be increased to 30–60mg a day in cases of hyponatremia <135mmol/l, or if the observed natremia increment is <5mmol/day. Since the vaptans correct hyponatremia by facilitating free water elimination, it is important to evaluate the patient blood volume and avoid their use in individuals with hypovolemic hyponatremia. These drugs therefore might be indicated in neurocritical patients with hyponatremia, though for the time being their administration should be limited to patients in which conventional therapy has failed, or as coadjuvant treatment.52
The recent European guidelines do not advise the use of these drugs in neurocritical patients with SIADH and moderate to severe hyponatremia.38 Lastly, these substances are contraindicated in patients with a glomerular filtration rate of <30ml/min, and in the presence of previous liver disease.53,54
Treatment of hyponatremia overcorrectionIn the case of accidental natremia overcorrection, with values of >10mmol/l during the first 24h, or of 8mmol/l during day 2, it is possible to reinduce hyponatremia (according to the guides of the European Society of Intensive Care Medicine/European Society of Endocrinology/European Renal Association-European Dialysis and Transplant Association)38 with the aim of avoiding central pontine myelinolysis. The following scheme can be used in cases of natremia overcorrection, particularly when the initial serum sodium value is 120mmol/l11,20,38:
The infusion of 3% HSS and of all treatments tending to reduce natriuresis should be interrupted.
The infusion of 5% dextrose solution (3ml/kg/h) as a means for administering electrolyte-free water is a useful management option.
The administration of intravenous desmopressin (2–4μg in 3 daily doses). It is important to mention that this drug is not indicated in patients with intracranial hypertension, due to the risk of increasing the brain edema.
Hourly monitoring of natremia with the purpose of avoiding a decrease beyond the predetermined target value.
ConclusionHyponatremia is the most frequent electrolytic disturbance in neurocritical patients, and is a leading cause of morbidity–mortality if adequate and immediate treatment is not provided. The syndrome of inappropriate antidiuretic hormone secretion and CSW have been described as the two syndromes that most often explain the presence of hyponatremia associated to increased natriuresis in neurocritical patients. In these cases the evaluation of volemia allows us to establish a differential diagnosis between the two disease conditions–though in many patients such differentiation is a genuine challenge. However, recent evidence has questioned the existence of CSW as a cause of hyponatremia in aneurysmal SAH. Conceptually, the diagnosis of cerebral salt wasting seems more appropriate for defining hyponatremia in the neurocritical patient, since the reference treatment is always the same, regardless on the underlying cause: the administration of hypertonic saline solution. Likewise, in the neurocritical ICU, the treatment of hyponatremia depends on a number of factors such as the underlying disease process, the speed with which the condition develops, and patient volemia. Lastly, some alternative treatments have been studied, such as fludrocortisone and the vaptans, though conclusive evidence regarding their use is still lacking, and no definitive agreement has been reached regarding their indications.
Conflicts of interestThe authors declare that they have no conflicts of interest.
Please cite this article as: Manzanares W, Aramendi I, Langlois PL, Biestro A. Hiponatremias en el paciente neurocrítico: enfoque terapéutico basado en la evidencia actual. Med Intensiva. 2015;39:234–243

