







Editado por: Federico Gordo - Medicina Intensiva del Hospital Universitario del Henares (Coslada-Madrid)
Última actualización: Octubre 2023
Más datosLos Enterobacterales resistentes a carbapenémicos o productores de betalactamasas de espectro extendido (BLEE) y los no fermentadores resistentes a carbapenémicos presentan resistencia a muchos de los antimicrobianos comúnmente empleados en la práctica clínica, y han sido reconocidos por la Organización Mundial de la Salud como una prioridad crítica para el desarrollo de nuevos antimicrobianos. En esta revisión se abordarán los principales mecanismos de resistencia de los Enterobacterales, Pseudomonas aeruginosa, Acinetobacter baumannii y Stenotrophomonas maltophilia a betalactámicos, quinolonas, aminoglucósidos y polimixinas. Se presentará información actualizada sobre la importancia en la resistencia de mecanismos de modificación de antimicrobianos (incluyendo betalactamasas de claseC de espectro extendido, carbapenemasas y enzimas modificadoras de aminoglucósidos), alteraciones de la permeabilidad por trastornos en la expresión de porinas o del lipopolisacárido, producción de bombas de expulsión activa, alteraciones de la diana o protección de la misma y expresión de sistemas de doble componente.
Enterobacterales resistant to carbapenems or producing extended-spectrum β-lactamases (ESBL) and non-fermenters resistant to carbapenems present resistance to many of the antimicrobials commonly used in clinical practice, and have been recognized by the World Health Organization as a critical priority for the development of new antimicrobials. In this review, the main mechanisms of resistance of Enterobacterales, Pseudomonas aeruginosa, Acinetobacter baumannii and Stenotrophomonas maltophilia to β-lactams, quinolones, aminoglycosides and polymyxins will be addressed. Updated information will be presented on the importance in resistance of antimicrobial modification mechanisms (including classC or extended-spectrum β-lactamases, carbapenemases and aminoglycoside-modifying enzymes), permeability alterations due to porin or lipopolysaccharide expression disorders, production of active efflux pumps, target alterations or protection, and expression of two-component systems.
La resistencia a los antimicrobianos es uno de los principales problemas de salud a nivel global, en especial cuando se consideran los microorganismos multirresistentes. La Organización Mundial de la Salud ha hecho pública una lista de prioridades en relación con bacterias para las que se necesitan con urgencia nuevos antimicrobianos, y en la misma se reconocen como de prioridad crítica a los Enterobacterales resistentes a carbapenémicos o productores de betalactamasas de espectro extendido (BLEE) y a los no fermentadores (Pseudomonas aeruginosa, Acinetobacter baumannii) resistentes a carbapenémicos1. Estos microorganismos, además, suelen presentar resistencia a otros grupos/familias de antimicrobianos comúnmente empleados en la práctica clínica. En este manuscrito se abordan, de forma sucinta, los principales mecanismos de resistencia de Enterobacterales, P.aeruginosa, A.baumannii y, también, Stenotrophomonas maltophilia.
EnterobacteralesResistencia a betalactámicosEl principal mecanismo de resistencia a betalactámicos en Enterobacterales es la producción de betalactamasas, enzimas que hidrolizan el anillo betalactámico impidiendo su actividad antibacteriana2,3. Atendiendo a su estructura molecular, se conocen cuatro grupos de betalactamasas (A, B, C yD)4. La actividad enzimática depende de un residuo de serina en las clases A, C yD, y de uno o dos iones de cinc en la claseB, por lo que estas últimas se denominan también metalobetalactamasas5. Por otra parte, las betalactamasas se pueden clasificar atendiendo a su capacidad para hidrolizar distintos sustratos y a su inhibición por diferentes compuestos en tres grupos funcionales: 1, 2 y 3 (tabla 1)6,7. Si se consideran aspectos clínicos, las enzimas de mayor interés en enterobacterias corresponden a tres grupos: BLEE, enzimas de claseC y carbapenemasas2,3.
Clasificación de betalactamasas de interés clínico
| Grupo funcionala | Clase molecular | Sustratos de referencia | Inhibición | Tipo de enzima | Ejemplos (familias/representantes) | |
|---|---|---|---|---|---|---|
| AC-TZB | EDTA | |||||
| 1 | C | Cefalosporinas | − | − | Cefalosporinasas cromosómicasCefamicinasas plasmídicas | AmpCsFOX, DHA, ACT… |
| 2a | A | Penicilinas | + | − | Penicilinasas | PC1 |
| 2b | A | PenicilinasCefalosporinas-1G | + | − | Penicilinasas de amplio espectro | TEM-1, SHV-1 |
| 2be | A | Cefalosporinas-EEMonobactámicos | + | − | BLEE | TEM, SHV, CTX-M… |
| 2br | A | Penicilinas | − | − | TEM resistentes a inhibidores | TEM-30… |
| 2ber | A | Cefalosporinas-EEMonobactámicos | − | − | BLEE resistentes a inhibidores | TEM-50… |
| 2c | A | Carbenicilina | + | − | Penicilinasas | PSE-1 |
| 2ce | A | CarbenicilinaCefepima | + | − | Penicilinasas-cefepimasas | RTG-4 |
| 2d | D | Oxacilina | ± | − | Oxacilinasas | OXA-1, OXA-10… |
| 2de | D | Cefalosporinas-EE | ± | − | Oxacilinasas de espectro extendido | OXA-11… |
| 2df | D | Carbapenémicos | ± | − | Carbapenemasas tipo OXA | OXA-23, OXA-48… |
| 2e | A | Cefalosporinas-EE | + | − | Carbapenemasas | CepA |
| 2f | A | Carbapenémicos | ± | − | Carbapenemasas | KPC, IMI, GES-6… |
| 3 | B | Carbapenémicos | − | + | Carbapenemasas | IMP, VIM, NDM…L1, CphA… |
AC/TZB: ácido clavulánico/tazobactam; BLEE: betalactamasas de espectro extendido; Cefalosporinas-EE: cefalosporinas de espectro expandido; Cefalosporinas-1G: cefalosporinas de primera generación.
Los grupos de especial importancia6,7 aparecen en negrita.
Las BLEE son enzimas de claseA que degradan penicilinas, cefalosporinas (salvo cefamicinas) y monobactámicos; con frecuencia están codificadas por genes plasmídicos8. Habitualmente se inhiben por ácido clavulánico, tazobactam, sulbactam y nuevos inhibidores7 (fig. 1). Desde un punto de vista estructural hay un amplísimo número de familias, siendo TEM, SHV (relacionadas con una enzima cromosómica intrínseca —sin perfil BLEE— de Klebsiella pneumoniae) y CTX-M (en particular CTX-M-15 y CTX-M-14) las más importantes9,10. Entre otros, los clones de Escherichia coli con tipo de secuencia (ST) ST131 o de K.pneumoniae ST11 y ST405 están implicados en la diseminación mundial de CTX-M-1511-13.
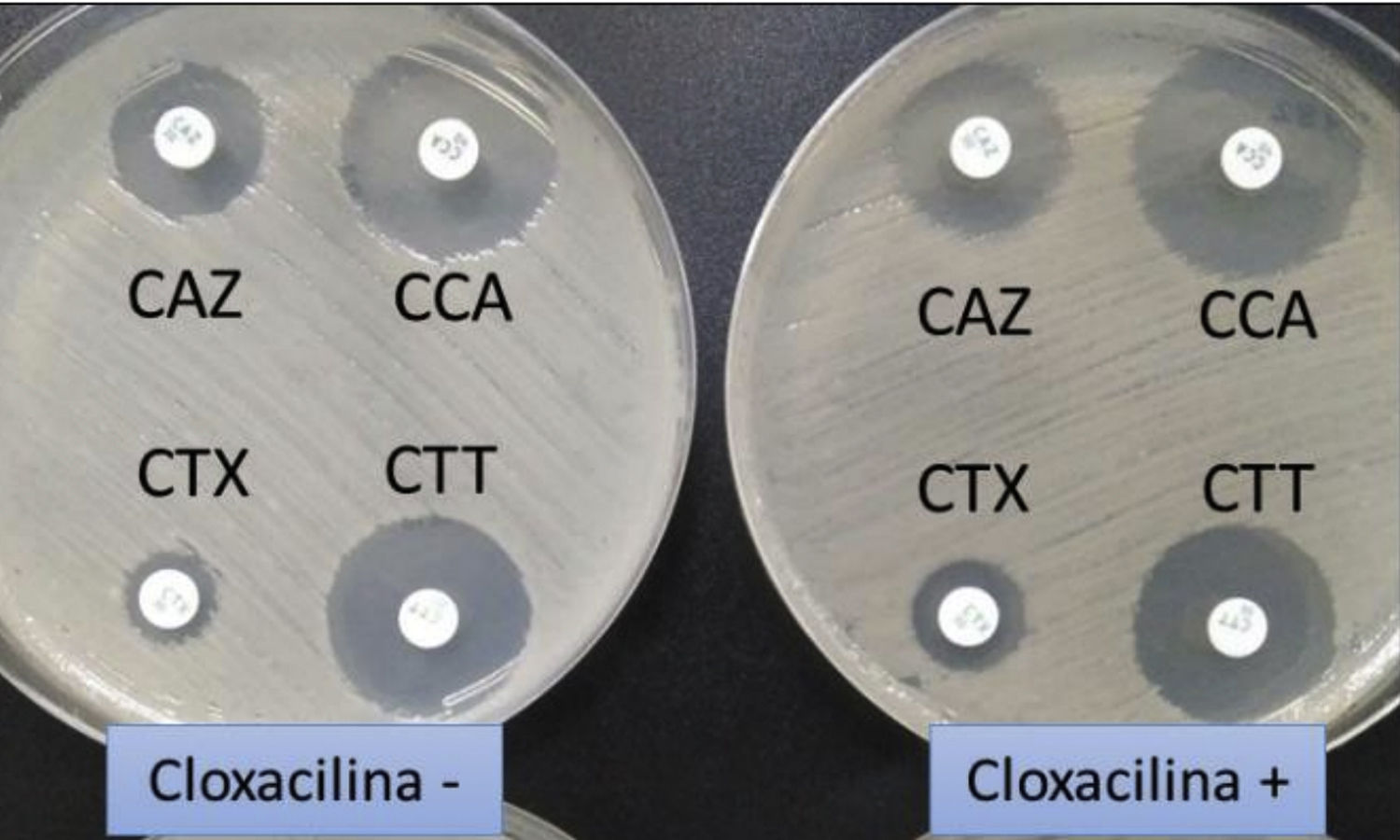
Klebsiella pneumoniae productora de betalactamasa de espectro extendido. En medio habitual (sin cloxacilina) se aprecia un menor diámetro de los halos de ceftazidima (CAZ) y cefotaxima (CTX) que de ambas cefalosporinas combinadas con ácido clavulánico (CCA y CTT, respectivamente). En medio con cloxacilina (que inhibiría la eventual presencia adicional de una betalactamasa —plasmídica en caso de K.pneumoniae— de claseC, no se observa un incremento de los halos, lo que descarta esa posibilidad.
Las betalactamasas de la clase molecularC incluyen tanto enzimas AmpC codificadas por genes cromosómicos14 como variantes codificadas por plásmidos (cefamicinasas plasmídicas)15. Como norma, no se inhiben por ácido clavulánico (ni tazobactam, ni sulbactam) pero sí por nuevos inhibidores como avibactam16. En muchas especies (no así en E.coli) la producción de AmpC cromosómica está regulada por un complejo sistema de genes; en condiciones basales, hay una baja producción de AmpC (estado de represión), pero ciertos betalactámicos pueden inducir con mayor o menor eficacia la producción de la enzima, y cuando desaparece el compuesto, cesa dicha inducción16,17. Las mutaciones en genes reguladores17 pueden determinar que se produzca una alta producción de enzima incluso si no hay betalactámico inductor, por lo que las cepas correspondientes se denominan desreprimidas18. La mayoría de las cefamicinasas plamídicas se producen a alto nivel15. El nivel de resistencia del microorganismo a cada betalactámico depende de la cantidad de enzima (capacidad de inducción del betalactámico o estado de desrepresión enzimática) y de la resistencia de cada compuesto a la hidrólisis enzimática14. Las enzimas de claseC pueden degradar penicilinas y cefalosporinas como cefotaxima y ceftazidima, y aunque no hidrolizan los carbapenémicos de forma eficaz (salvo alguna excepción, como CMY-10 o ACT-28), cuando la enzima se hiperproduce en cepas con mecanismos adicionales (ver más adelante) puede alcanzarse un nivel de resistencia de importancia clínica19.
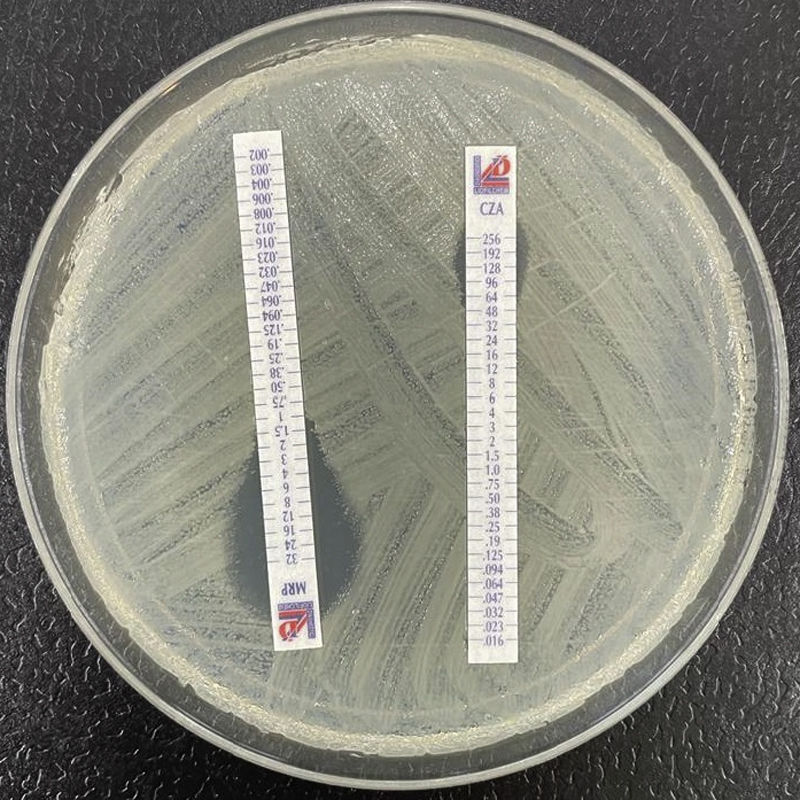
Las betalactamasas con actividad carbapenemasa de claseA tienen como principales representantes a las de la familia KPC, y en menor medida GES (no todas sus variantes tienen actividad carbapenemasa), SME, IMI y otras variantes20-22. Las KPC tienen distribución universal y, genéricamente, hidrolizan carbapenémicos, penicilinas, cefalosporinas y monobactámicos; no se inhiben con ácido clavulánico (de hecho, lo hidrolizan) pero sí por avibactam, vaborbactam y relebactam. Se conocen hasta ahora casi 100 variantes de KPC, varias de las cuales (p.ej., KPC-31) ocasionan resistencia a ceftazidima-avibactam pero no hidrolizan eficientemente los carbapenémicos, generando así un fenotipo similar al de las BLEE23 (fig. 2). El gen blaKPC forma parte del transposón Tn4401 (del que existen varias isoformas) y se vehicula por diversos plásmidos que también codifican genes de resistencia a otras familias de antimicrobianos. Se han identificado diversas especies de enterobacterias productoras de KPC, pero tienen especial importancia, por su frecuencia, los denominados clones de alto riesgo de K.pneumoniae ST258, ST14, ST15, ST307, etc.24.
Las betalactamasas de claseB, o metalobetalactamasas, degradan los carbapenémicos y otros compuestos (penicilinas, cefalosporinas, pero no monobactámicos) por un mecanismo dependiente de Zn2+. Estas enzimas no se inhiben con los inhibidores actualmente disponibles. Aunque sí se inhiben con EDTA (u otros quelantes), ello no tiene utilidad desde el punto de vista terapéutico25. De las diferentes familias conocidas, en enterobacterias tienen una amplia diseminación geográfica las NDM, de codificación plasmídica o cromosómica, en aislados de distintas especies (E.coli ST101 o ST131, K.pneumoniae…). También se han descrito cepas productoras de VIM, IMP u otras familias.
Las enzimas de claseD se conocen genéricamente como oxacilinasas porque hidrolizan in vitro este compuesto más eficientemente que las enzimas de las clasesA oC26,27. Estructuralmente, son un grupo heterogéneo de betalactamasas que pueden comportarse como penicilinasas, betalactamasas de espectro extendido y carbapenemasas. Salvo excepciones, no se inhiben con ácido clavulánico, tazobactam o sulbactam; algunas de ellas (p.ej., grupo de OXA-48) se inhiben con avibactam. En Enterobacterales, la carbapenemasa más relevante de entre las oxacilinasas es OXA-48, inicialmente identificada en K.pneumoniae en Turquía pero con amplia diseminación en la cuenca mediterránea y en otros países. Habitualmente, blaOXA-48 se asocia a un plásmido conjugativo y a variantes de Tn1999 encontrados en diferentes ST de K.pneumoniae y otras especies. Aunque OXA-48 tiene poca capacidad de hidrólisis de cefalosporinas de espectro expandido, muchas cepas con esta enzima suelen producir también CTX-M-15, por lo que presentan resistencia a dichos compuestos28. Se han otras descrito variantes relacionadas con OXA-48, como OXA-163 (con actividad frente a cefalosporinas de espectro expandido) u OXA-181.
Además de la producción de betalactamasas, la resistencia a betalactámicos en Enterobacterales se relaciona con otros mecanismos. La pérdida o la alteración estructural de las porinas (canales hidrófilos a través de los cuales los antimicrobianos alcanzan el interior de la bacteria) causan por sí mismas un pequeño incremento de resistencia que, en ausencia de otros mecanismos, tendría un limitado impacto clínico, pero que sí actúa sinérgicamente con ellos incrementando la resistencia. El impacto es mayor cuando se pierden en una misma cepa sus distintas porinas: muchos clones de K.pneumoniae productores de BLEE o de carbapenemasas carecen ya de la porina OmpK35, por lo que la pérdida adicional de la segunda porina principal (OmpK36) causa un marcado incremento de la resistencia29. La pérdida de porinas es también relevante en otras especies, como Enterobacterspp.19; curiosamente, en E.coli, aunque se conocen múltiples detalles del papel de las porinas en la resistencia en la cepa de laboratorio K-12, disponemos de menos información cuando se consideran aislamientos clínicos. Las bombas de expulsión activa, que eliminan betalactámicos una vez que estos han penetrado en la bacteria, también contribuyen al aumento de la resistencia, pero su papel en el caso de los betalactámicos de mayor interés clínico es menos relevante que en comparación con otros antimicrobianos o con el que juegan en otros microorganismos (como se detalla al tratar de P.aeruginosa)30. La combinación de pérdida de porinas, hiperproducción de la bomba AcrB y la hiperexpresión de KPC-2 se ha relacionado con la resistencia a la nueva combinación meropenem-vaborbactam.
A diferencia de lo que ocurre en bacterias grampositivas como Staphylococcus aureus o Streptococcus pneumoniae, el papel de las alteraciones de las proteínas fijadoras de penicilina (PBP, por sus iniciales en inglés) en la resistencia de los Enterobacterales a betalactámicos es pequeño, aunque algunos estudios han indicado el interés de mutaciones en la PBP-331.
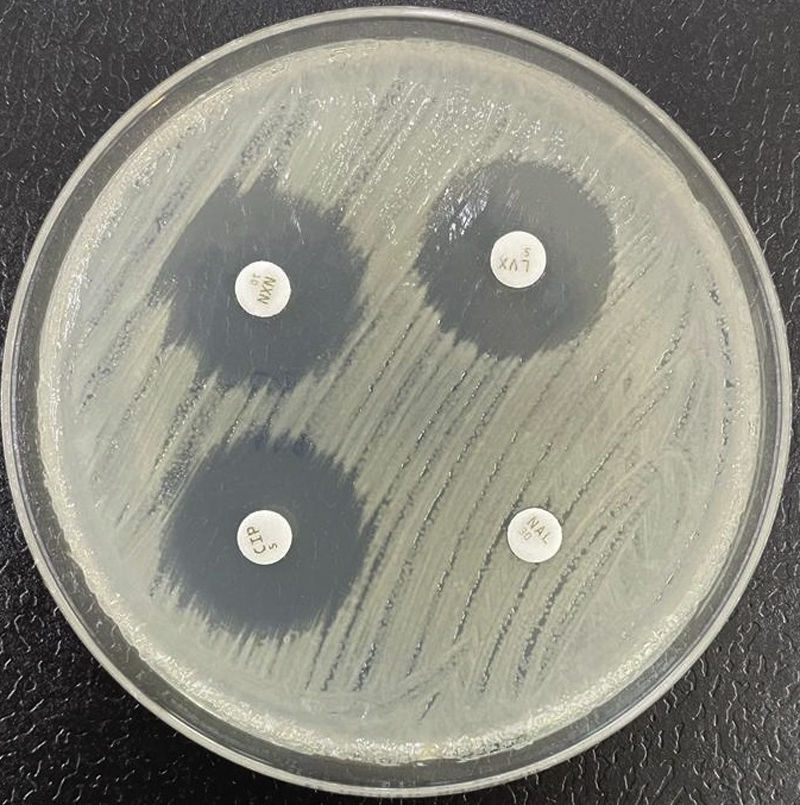
Resistencia a quinolonasLa resistencia a quinolonas en Enterobacterales se relaciona con múltiples mecanismos. Tradicionalmente, se ha prestado especial atención a la resistencia derivada de alteraciones en las topoisomerasas de tipoII (topoisomerasaII o ADN girasa y topoisomerasaIV). Estas ocurren como consecuencia de mutaciones en la llamada «región determinante de resistencia a quinolonas» (QRDR, por sus iniciales en inglés) de los genes gyrA (ADN-girasa) y parC (topoisomerasaIV). Menos importantes son las mutaciones en gyrB o parE. Una mutación única en gyrA causa incremento de la resistencia a las quinolonas no fluoradas (ácido nalidíxico) y bajo nivel de resistencia a las fluoradas (fig. 3), pero el nivel de resistencia a esta últimas se incrementa de forma paralela al del número de nuevas mutaciones que ocurran en gyrA y parC32.
Las alteraciones en las porinas (como en el caso de los betalactámicos) contribuyen al aumento del nivel de resistencia a quinolonas. Se ha comprobado también que las modificaciones del lipopolisacárido guardan relación con resistencia a estos compuestos. En relación con las bombas de expulsión activa, son de especial interés las de la familia RND (resistencia-nodulación-división) que se integran estructuralmente en sistemas de tres proteínas: la propia bomba, un canal de expulsión en la membrana externa y una proteína acopladora de las otras dos. Los sistemas RND eliminan múltiples compuestos, entre ellos quinolonas. El ejemplo mejor estudiado es el de AcrA-AcrB-TolC, cuya expresión contribuye al (bajo) nivel basal de resistencia30; este sistema se puede hiperexpresar por mutaciones en otros genes reguladores (operón mar, acrR…), aumentado, en consecuencia, el nivel de resistencia. Algunos de estos reguladores (mar) son capaces de conducir simultáneamente a la pérdida de una porina y a la hiperproducción de la bomba de expulsión33. La gran mayoría de cepas de K.pneumoniae poseen los genes cromosómicos oqxAB, que codifican un mecanismo de expulsión activa de quinolonas hidrófilas y otros compuestos.
Se han identificado varias familias de proteínas Qnr34 codificadas por plásmidos que causan resistencia a quinolonas por un mecanismo de protección de la diana de estos compuestos (topoisomerasas). Incluso, en varias especies, se han identificado genes cromosómicos intrínsecos de tipo qnr. Tras el descubrimiento de Qnr, se han identificado otros mecanismos de resistencia plasmídica relacionados con la producción de la acetilasa AAC(6’)-Ib-cr, que afecta tanto a (algunas) quinolonas como a (algunos) aminoglucósidos o con la expresión QepA (bomba de expulsión activa). Estos mecanismos plasmídicos causan per se (muy) bajo nivel de resistencia, pero cuando se acumulan en la misma cepa o coinciden con mecanismos cromosómicos, el nivel sobrepasa los puntos de corte clínicos de resistencia35.
Resistencia a aminoglucósidosLos dos mecanismos fundamentales de resistencia a aminoglucósidos en Enterobacterales son la modificación enzimática de estos compuestos o la modificación de su diana36,37. También disponemos de información adicional sobre trastornos en la penetración en la bacteria o la eliminación activa por bombas de expulsión (AcrD)30.
Las enzimas modificadoras de aminoglucósidos se agrupan en tres grandes familias de nucleotidil (adenil)-transferasas, fosfo-transferasas o acetil-transferasas, con infinidad de variantes cada una, que transfieren a ciertas posiciones de las moléculas de aminoglucósidos AMP, fosfato o acetil-coenzimaA, respectivamente, anulando su efecto antibacteriano36.
Cada enzima concreta afecta a ciertos aminoglucósidos, pero no a otros. Además, un mismo microorganismo puede producir múltiples enzimas de igual o diferente familia, por lo que los fenotipos de resistencia son difíciles de correlacionar con proteínas (genes) singulares. Para mayor complejidad, hay dos sistemas diferentes de nomenclatura de estas enzimas (para las proteínas y para sus genes). Las enzimas más frecuentemente halladas en Enterobacterales (la mayoría de estudios se refieren a E.coli y K.pneumoniae multirresistentes) suelen ser AAC(3)-IIa, AAC(3)-IVa, AAC(6’)-Ib (con variantes que afectan a las quinolonas, como se ha reseñado antes), ANT(2”)-Ia, APH (3’)-Ia, APH(3’)-IIa, APH(3”)-Ib, ANT(2”)-Ia y ANT(3”)-Ia; debe tenerse en cuenta, en todo caso, que ciertas enzimas afectan a compuestos que, en la actualidad, tienen escaso interés clínico (p.ej., ANT3”-Ia afecta solamente a estreptomicina y espectinomicina)36. El nuevo compuesto plazomicina escapa a la práctica totalidad de este tipo de enzimas modificadoras38.
La modificación ribosómica como causa de resistencia a aminoglucósidos puede deberse a alteraciones en las proteínas del ribosoma o —más importante por su frecuencia— a la modificación de sitios específicos en el 16SrRNA por metil-transferasas (metilasas) de codificación plasmídica37. Hay dos grandes familias de estas metilasas: N7-G1405 (ArmA y variantes de Rmt) y N1-A1408 (NmpA). Las dos familias inactivan los aminoglucósidos de interés clínico (gentamicina, tobramicina, amikacina, e incluso plazomicina), ocasionando resistencia de alto nivel, y se diferencian entre sí por inactivar o no otros compuestos que raramente se usan en la práctica clínica actual. Mucha de la información disponible sobre este mecanismo de resistencia se refiere al estudio de cepas multirresistentes; en España, recientemente, se ha comprobado que el 5,1% de enterobacterias productoras de carbapenemasa (principalmente, K.pneumoniae, E.cloacae) producen metilasas, en particular RmtF39.
Resistencia a polimixinasVarias de las enterobacterias más frecuentemente aisladas en muestras clínicas presentan resistencia intrínseca a polimixinas, incluyendo Proteusspp., Morganella, Providenciaspp., Serratiamarcescens y Hafnia alvei.
Otras especies pueden desarrollar resistencia adquirida por diversos mecanismos, de los que el más importante es la modificación del lipopolisacárido (LPS) por la adición de diversas moléculas, más frecuentemente 4-amino-4-deoxi-L-arabinosa o galactosamina (mediada por genes cromosómicos) o fosfoetanolamina (por genes cromosómicos, o por los recientemente descubiertos genes plasmídicos de tipo mcr)40-42. Tras esta modificación, la carga negativa neta del LPS disminuye, con lo que se dificulta la interacción de las polimixinas con la bacteria. La importancia comparativa de otros mecanismos, como la producción de cápsula o bombas de expulsión activa, es secundaria.
Los genes cromosómicos que añaden los compuestos indicados se activan como consecuencia de mutaciones en sistemas de doble componente (constituidos por una proteína histidina-quinasa transmembrana sensora que se autofosforila en determinadas condiciones, y otra proteína citoplásmica que, cuando se fosforila, modula la expresión de distintos genes). Entre estos sistemas se incluyen PhoP-PhoQ, PmrA-PmrB y CrrA-CrrB40-42. En K.pneumoniae, las mutaciones de MgrB (una pequeña proteína transmembrana que en condiciones normales regula negativamente la actividad quinasa del sistema PhoP/PhoQ) son una de las principales causas de resistencia a polimixinas43.
La familia de genes plasmídicos mcr codifica transferasas de fosfoetanolamina que modifican el LPS de forma análoga a como hacen otros genes cromosómicos. Su expresión no siempre se traduce en niveles que sobrepasan el punto de corte clínico de resistencia41.
Bacilos gramnegativos no fermentadoresPseudomonas aeruginosaResistencia a betalactámicosP.aeruginosa posee una cefalosporinasa cromosómica de tipo AmpC (fig. 4), y su expresión constitutiva caracteriza la resistencia natural de P.aeruginosa a las aminopenicilinas (incluida la asociación amoxicilina-clavulánico), cefalosporinas de primera y segunda generación, algunas cefalosporinas de tercera generación (cefotaxima, ceftriaxona) y ertapenem44. Las ureidopenicilinas (piperacilina, ticarcilina), algunas cefalosporinas de tercera generación (ceftazidima) y la cefalosporina de cuarta generación cefepima, no se afectan por la expresión basal de esta betalactamasa.
Penicilinas y cefalosporinas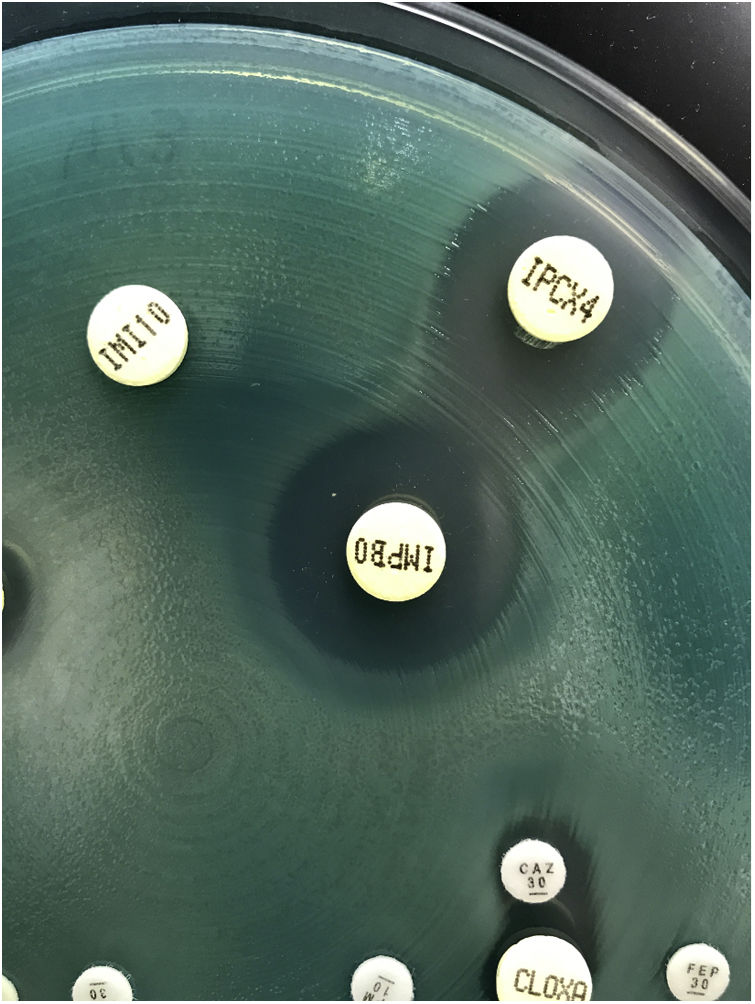
P.aeruginosa posee una importante capacidad para desarrollar resistencia por mutación, principalmente la selección de mutantes con hiperproducción constitutiva de su cefalosporinasa cromosómica AmpC45, siendo este el principal mecanismo de resistencia a las penicilinas (piperacilina, piperacilina-tazobactam) y cefalosporinas (ceftazidima y cefepima). La hiperproducción unida a modificaciones estructurales de la cefalosporinasa cromosómica AmpC es el principal mecanismo de resistencia clínica a ceftolozano/tazobactam y ceftazidima/avibactam in vivo46,47.
Por otro lado, la hiperexpresión de cualquiera de sus múltiples bombas de expulsión, principalmente MexAB-OprM, MexXY-OprM y MexCD-OprJ, contribuye significativamente a los fenotipos de resistencia a cefalosporinas. La hiperexpresión mutacional de MexAB-OprM afecta por igual a cefepima y a ceftazidima, y la de MexXY-OprM y MexCD-OprJ, a cefepima48.
P.aeruginosa puede también presentar resistencia a las cefalosporinas mediada por betalactamasas de espectro extendido (BLEE) vehiculizadas en plásmidos y/o integrones que afectan a la sensibilidad de ceftazidima y de cefepima, pero no de manera uniforme. Las betalactamasas tipo GES, PER, TEM, SHV y VEB tienen actividad ceftazidimasa principalmente, afectando en menor medida a cefepima, y algunas BLEE del tipo GES pueden afectar a los carbapenémicos49. Las BLEE tipo OXA pueden presentar, según el tipo, actividad ceftazidimasa o cefepimasa50, y recientemente se ha descrito el desarrollo de resistencia mediada por OXA-10 a ceftolozano/tazobactam y ceftazidima/avibactam51.
CarbapenémicosLa resistencia a los carbapenémicos en P.aeruginosa se asocia, generalmente, a mutaciones cromosómicas que alteran sus porinas, la sobreexpresión de bombas de expulsión, la desrepresión de betalactamasas intrínsecas o una combinación de ellas52. Pero estos mecanismos no afectan de manera uniforme a imipenem y a meropenem.
El principal mecanismo de resistencia al imipenem es la represión o inactivación de la porina OprD, que, junto con la expresión inducible de su cefalosporinasa AmpC, aumenta su CMI basal a valores de 8-32mg/l53 (fig. 3). Meropenem, sin embargo, aunque también utiliza OprD, se sirve de otras vías alternativas de entrada, de manera que experimenta un aumento más discreto de la CMI con valores de 2-4mg/l54. La hiperexpresión de la bomba MexAB-OprM más la inactivación de OprD es la causa más frecuente de resistencia clínica a meropenem55.
En menor medida, la resistencia a carbapenémicos puede ser mediada por carbapenemasas adquiridas, principalmente metalobetalactamasas, que son capaces de hidrolizar la mayoría de los antibióticos betalactámicos. Los genes que codifican estas metalobetalactamasas generalmente se encuentran en integrones que con frecuencia llevan genes adicionales que codifican la resistencia a antibióticos no betalactámicos. Las metalobetalactamasas más prevalentes y extendidas son los tipos VIM e IMP, y en menor medida NDM56 (fig. 5). Como se destacó anteriormente, algunas betalactamasas BLEE del tipo GES muestran actividad carbapenemasa.
Resistencia a fluoroquinolonas
La resistencia de alto nivel a fluoroquinolonas es debida a la interacción de mutaciones en las topoisomerasas, incluyendo ADN girasa (GyrA y GyrB) y topoisomerasaIV (ParC y ParE) asociada a la hiperexpresión por mutación de las bombas de expulsión MexAB/XY/CD/EF49.
La resistencia de bajo nivel a las quinolonas generalmente está mediada por plásmidos y generalmente asociada a enzimas modificadoras AAC(6’)-Ib-cr57.
Resistencia a aminoglucósidosLa resistencia a aminoglucósidos generalmente viene mediada por tres mecanismos: modificación enzimática, mecanismos de expulsión activa y metilación del ARN16s.
Los mecanismos de modificación enzimática están codificados frecuentemente por plásmidos, pudiendo ser de tres tipos: acetiltransferasas (AAC), adeniltransferasas (ANT) y fosforiltransferasas (APH). Estas enzimas, no afectan de manera uniforme a todos aminoglucósidos. Entre las más prevalentes se encuentran AAC(69)-II, AAC(3)-II y ANT(29)-I, que determinan resistencia a gentamicina y a tobramicina, mientras que AAC(3)-I está asociada con resistencia a gentamicina58.
Otros mecanismos incluyen la reducción de la concentración intracelular de aminoglucósidos por cambios en la permeabilidad de la membrana externa o bombas de expulsión (MexAB-OprM)59 y mecanismos de metilación del ARN16s mediados por metilasas codificadas en transposones insertados en plásmidos, que confieren una resistencia de alto nivel a todos los aminoglucósidos60.
Adicionalmente, se describen aumentos graduales en la CMI de los aminoglucósidos debido a mecanismos no enzimáticos asociados a mutación en su lipopolisacárido o proteínas externas de membrana.
Resistencia a polimixinasEl desarrollo de resistencia a colistina generalmente implica la modificación de su lipopolisacárido mediada por mutaciones en los sistemas de dos componentes codificados por genes pmrAB, phoPQ o parRS61.
De manera similar, la expresión inducible del operón arnBCADTEF, responsable de la adición de un 4-aminoarabinosa residual al lípidoA del lipopolisacárido, es fundamental para el desarrollo de resistencia inducible y/o adaptativa a la colistina62.
Acinetobacter baumanniiResistencia a betalactámicos- •
Cefalosporinas. A.baumannii posee una sensibilidad reducida a las cefalosporinas, debido a que produce una cefalosporinasa de tipo AmpC no inducible y una oxacilinasa de tipo OXA-51, lo que, unido a una expresión constitutiva de bajo nivel de una o más de sus bombas de expulsión63, confiere resistencia intrínseca a cefalosporinas de primera, de segunda generación y a algunas cefalosporinas de tercera generación, como cefotaxima y ceftriaxona64. La resistencia al resto de cefalosporinas de amplio espectro puede estar mediada por la sobreexpresión de estos mecanismos de resistencia intrínsecos. La sobreexpresión de su cefalosporinasa AmpC intrínseca por una secuencia de inserción ISAba-1 conduce a resistencia de alto nivel a ceftazidima y cefepima65. La sobreexpresión por mutación de la bomba de expulsión AdeABC, también confiere resistencia de alto nivel a cefalosporinas66.
Por otro lado, la resistencia a cefalosporinas puede estar mediada por la adquisición de betalactamasas plasmídicas, principalmente del tipo PER, VEB, GES, aunque también de BLEE tipo TEM y SHV67. Este mecanismo produce resistencia de alto nivel a ceftazidima y cefepima.
- •
Carbapenémicos. La resistencia a los carbapenémicos en A.baumannii se relaciona con numerosas betalactamasas con actividad carbapenemasa, incluidas carbapenemasas de tipo OXA, tanto constitutivas, como OXA-51/69 (hiperproducida por la inserción de la secuencia ISAba1), o bien adquiridas, principalmente oxacilinasas del grupo OXA-23, del grupo OXA-24/40, OXA-58 y OXA-143, metalobetalactamasas tipo IMP, VIM, SIM y NDM o betalactamasas de espectro extendido (BLEE) tipo GES68. Estos mecanismos conducen a resistencia de alto nivel a imipenem y a meropenem con valores de CMI en el rango de 16-32mg/l. A veces, la resistencia está asociada con la expresión disminuida de sus proteínas de membrana externa.
Además, la sobreexpresión del gen adeB, regulado por los genes adeRS (sistema regulador de dos componentes de la familia de bombas de expulsión AdeABC), también contribuye a la resistencia a carbapenémicos, aumentando alrededor de dos veces el valor de la CMI de imipenem y de meropenem69.
Resistencia a quinolonasLa resistencia a fluoroquinolonas es generalmente el resultado de mutaciones cromosómicas que afectan a las regiones determinantes de resistencia a quinolonas de la ADN girasa (GyrA y GyrB) y topoisomerasaIV (ParC y ParE), lo que conduce secuencialmente a resistencia de alto nivel a ciprofloxacino y levofloxacino70. Además, la resistencia también puede estar mediada por bombas de expulsión, como AdeABC y AdeM71, que incrementan la CMI a niveles más bajos. En este caso, no solo las fluoroquinolonas sino también los aminoglucósidos y las tetraciclinas son afectados.
Resistencia a aminoglucósidosLa resistencia puede estar mediada por diversos mecanismos: a)bombas de expulsión; b)alteraciones en la diana, y 3)enzimas modificantes de los aminoglucósidos72. Entre las bombas de expulsión, la principal es AdeABC (también implicada en la resistencia a quinolonas), que afecta a la sensibilidad a gentamicina, a tobramicina y a amikacina. Una segunda bomba, AbeM, afecta principalmente a gentamicina.
La producción de metilasas del ARN16s está mediada por plásmidos; la principal es la ArmA, que confiere alto nivel de resistencia a gentamicina, a tobramicina y a amikacina (CMI>256mg/l). Respecto a las enzimas modificantes de aminoglucósidos, están mediadas por acetiltransferasas plasmídicas, nucleotidiltransferasas y/o fosfotransferasas, solas o en combinación, principalmente la familia AAC(6́), que en sus diversas variantes afecta a gentamicina, a tobramicina y/o a amikacina (y que conducen a resistencia de alto nivel (CMI≥32mg/l).
Resistencia a tetraciclinasLa resistencia a tetraciclinas de primera generación (tetraciclina), segunda generación (doxiciclina, minociclina) y tigeciclina (análogo estructural de minociclina) está mediada por bombas de expulsión.
La resistencia a la primera y segunda generación de tetraciclinas se asocia generalmente a bombas de expulsión codificadas por los genes tetA y tetB localizados en transposones. El gen tetA es responsable de la resistencia a tetraciclina y a doxiciclina, pero no a minociclina, mientras que el gen tetB se ha encontrado en los aislamientos que también eran resistentes a la minociclina73.
La resistencia a tigeciclina se asocia principalmente a la hiperexpresión de la bomba de expulsión AdeABC; otra bomba de expulsión (AdeIJK) podría actuar sinérgicamente con AdeABC74.
Resistencia a polimixinasSe han descrito dos mecanismos de resistencia a la colistina en A.baumannii: a)alteraciones en el lípidoA del LPS como resultado de mutaciones en el sistema de dos componentes PmrAB75, y b)pérdida completa de la producción de LPS resultante de mutaciones en los genes lpxA, lpxC y lpxD que codifican las enzimas que catalizan los primeros pasos en la biosíntesis de LPS76. La resistencia a colistina mediada por el gen mrc de transmisión plasmídica no ha sido descrita en A.baumannii77.
Stenotrophomonas maltophiliaS.maltophilia presenta un fenotipo intrínseco de multirresistencia que está relacionado con la baja permeabilidad de su membrana externa debido a su bajo número de porinas78 y a la presencia de una bomba de expulsión SmeDEF, que afecta a betalactámicos, a quinolonas y a aminoglucósidos79. Además, es naturalmente resistente a los aminoglucósidos, debido a la presencia de una acetiltransferasa cromosómica, AAC(6’)-Iz80. También produce dos betalactamasas cromosómicas inducibles: la carbapenemasa L1, que confiere resistencia intrínseca a todos los carbapenémicos, y una cefalosporinasa L2, que, además, hidroliza aztreonam78.
Las fluoroquinolonas, y en particular el levofloxacino, son activas a pesar de la expresión a bajo nivel de una proteína Qnr codificada a nivel cromosómico. La resistencia de alto nivel a las fluoroquinolonas puede aparecer por la selección de mutantes con una mayor expresión del gen smQnr o de la bomba de expulsión SmeDEF81.
Aunque S.maltophilia es muy sensible a la combinación trimetoprim-sulfametoxazol, la resistencia a la combinación puede aparecer por la adquisición de genes sul y dfrA, vehiculizados por plásmidos e integrones de clase1, que codifican dihidropteroato sintetasas con actividad sobre las sulfonamidas82.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

